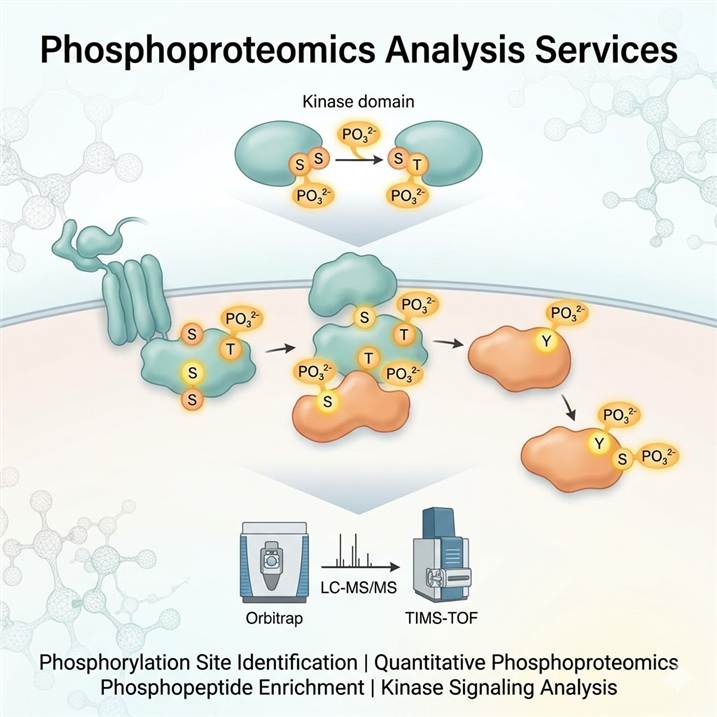
Meeting the Challenge of Phosphorylation Analysis
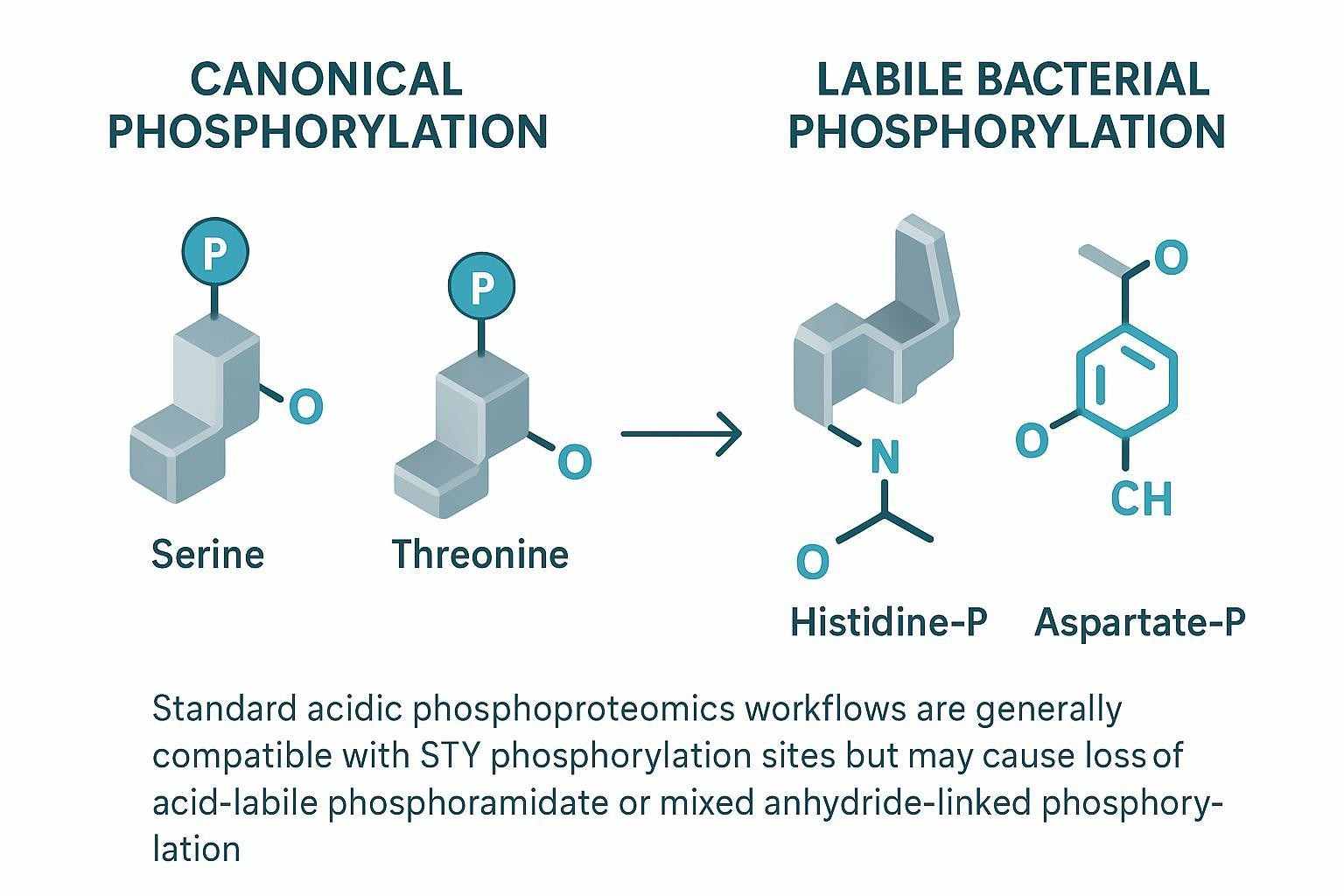
Protein phosphorylation is a reversible, dynamic modification that regulates virtually every aspect of cellular function. Unlike many PTMs with relatively uniform mass shifts, phosphorylation analysis presents distinct challenges: phosphorylation is often substoichiometric (only a small fraction of a protein may be phosphorylated at any given time), the phosphoester bond is labile under standard mass spectrometry conditions, and the unambiguous assignment of phosphorylation to a specific serine, threonine, or tyrosine residue requires specialized fragmentation strategies. Addressing these challenges demands dedicated analytical workflows optimized at every stage — from phosphopeptide enrichment through mass spectrometry acquisition to site localization scoring.
Why Phosphoproteomics Demands a Dedicated Approach
The dynamic and substoichiometric nature of protein phosphorylation means that phosphopeptides are vastly outnumbered by non-phosphorylated peptides in a typical digest. Without effective enrichment, most phosphorylation events escape detection entirely. Even after enrichment, confident site localization requires fragmentation methods that generate sequence-informative ions while preserving the phosphate group on the modified residue. Traditional collision-based fragmentation can cause neutral loss of phosphoric acid, compromising site assignment. Our platform addresses these challenges with optimized IMAC and MOAC enrichment chemistries, electron-based fragmentation methods, and advanced site localization algorithms.
Our phosphoproteomics platform integrates multiple strategies — from broad phosphoproteome discovery to targeted phosphorylation site validation — allowing researchers to select the analytical depth that matches their biological question. Whether mapping global signaling networks in drug-treated cells, profiling tyrosine phosphorylation in cancer tissues, or quantifying specific phosphorylation events in a time-course experiment, our services deliver the site-resolved data needed for confident biological interpretation. For broader integration with other PTM types, our PTM Crosstalk Analysis service provides multi-modification co-regulation insights.
Our Phosphoproteomics Service Portfolio
We offer a structured portfolio of phosphoproteomics services designed to address specific research objectives — from global phosphoproteome surveys to targeted quantification of individual phosphorylation sites. The table below maps common research goals to our recommended service modules.
| Research Objective |
Recommended Service |
Key Technology |
| Global phosphorylation site identification and phosphoproteome discovery |
Phosphorylation Site Identification |
IMAC/TiO₂ enrichment, HCD/EThcD, high-resolution Orbitrap MS |
| Quantitative comparison of phosphorylation across conditions |
Quantitative Phosphoproteomics Analysis |
TMT labeling, SILAC, label-free quantification, MRM/PRM |
| Specialized bioinformatics and phosphoproteomics data interpretation |
Phosphoproteomics Data Analysis |
MaxQuant, Philosopher, kinase-substrate prediction, motif analysis |
Each service module is available independently, or services can be combined into an integrated phosphorylation characterization workflow. For kinase drug discovery applications, our related Kinase Activity Profiling and Kinase-Substrate Network Analysis services provide complementary functional and systems-level perspectives on phosphorylation signaling.
Integrated Technical Platform for Phosphoproteomics
Reliable phosphoproteomics data depends on optimized methods across the entire analytical pipeline — from phosphopeptide enrichment through mass spectrometry acquisition to computational site localization. Our platform integrates best-in-class approaches at each stage, configurable to match the complexity and objectives of your project.
Phosphopeptide Enrichment Strategies
Phosphopeptide enrichment is the critical first step that determines the depth and quality of phosphoproteomic analysis. We deploy multiple complementary enrichment strategies, selected and combined based on sample type and research objective. IMAC (immobilized metal affinity chromatography) using Fe³⁺ or Ti⁴⁺ provides broad phosphopeptide coverage and is our default for global phosphoproteomics. MOAC (metal oxide affinity chromatography) using TiO₂ offers complementary selectivity, particularly for multiply phosphorylated peptides. For targeted analysis of specific phosphorylation types, antibody-based enrichment (e.g., anti-phosphotyrosine) enables deep coverage of low-abundance phosphorylation events. Sequential enrichment strategies combining IMAC and TiO₂ are used to maximize phosphoproteome depth. Our Phosphopeptide Enrichment service provides detailed information on enrichment strategy selection and optimization.
High-Resolution LC-MS/MS Acquisition
Phosphopeptide analysis is performed on high-resolution Orbitrap platforms, optimized for the distinct fragmentation behavior of phosphorylated peptides. Nano-flow LC configurations maximize sensitivity for limited sample amounts, while capillary-flow and micro-flow systems provide throughput for larger cohort studies. Data-dependent acquisition with dynamic exclusion is the standard for discovery phosphoproteomics, with scheduled PRM methods developed for targeted site quantification. For deep phosphoproteome coverage, we employ multi-injection fractionation strategies (high-pH reversed-phase fractionation, SAX, or SCX) to reduce sample complexity and increase the number of identified phosphorylation sites.
Multi-Mode Fragmentation for Site Localization
Confident phosphorylation site localization requires fragmentation methods that produce sequence-informative ions while preserving the phosphate moiety. We employ a multi-mode strategy tailored to the research objective. Stepped HCD provides robust peptide identification with neutral loss information that aids phosphosite localization. EThcD (electron-transfer/higher-energy collision dissociation) is the preferred method for unambiguous phosphorylation site assignment, as it preserves the phosphate group during peptide backbone fragmentation and generates comprehensive c- and z-type fragment ions. For phosphotyrosine analysis, higher-energy C-trap dissociation with supplemental activation provides enhanced sequence coverage and confident localization. Phosphosite localization confidence is scored using established algorithms including MaxQuant localization probability scores and PhosphoRS.
Quantitative Phosphoproteomics Strategies
Phosphorylation is inherently dynamic, and quantitative information is essential for biological interpretation. Our platform supports multiple quantification strategies. Label-free quantification using extracted ion chromatogram alignment is suitable for discovery studies and pilot experiments. TMT labeling enables multiplexed comparison of up to 16 conditions within a single experiment, reducing missing data across runs. SILAC provides metabolic labeling for cell culture models with maximum accuracy. For targeted validation of specific phosphorylation sites, MRM and PRM methods deliver absolute quantification with synthetic phosphopeptide standards.
Phosphoproteomics Workflow: From Sample to Publication-Ready Data
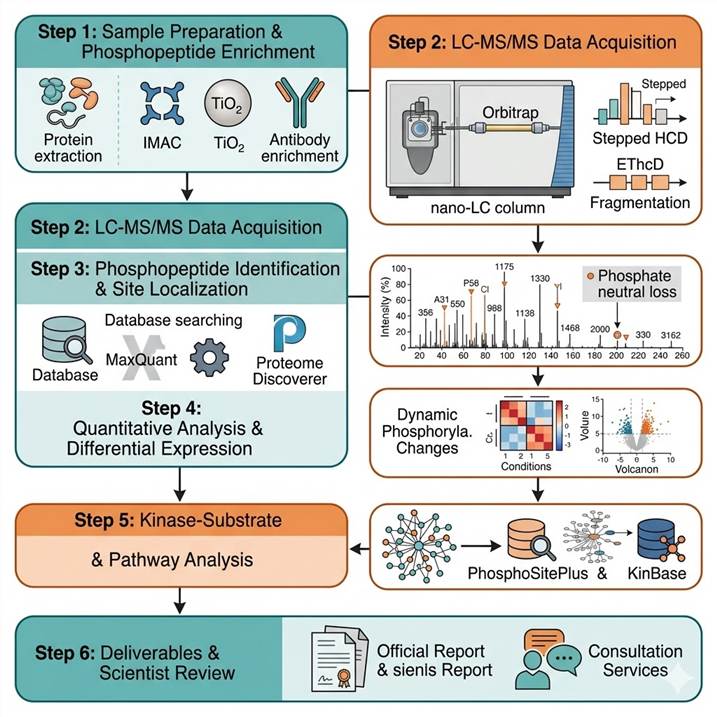
Step 1: Sample Preparation and Phosphopeptide Enrichment
Proteins are extracted under conditions that preserve endogenous phosphorylation (phosphatase inhibitors, cold temperature). Samples are reduced, alkylated, and digested using optimized proteases. Phosphopeptides are enriched using IMAC, TiO₂, or antibody-based methods selected based on sample type and research objective. Sequential enrichment strategies are applied for maximal phosphoproteome coverage.
Step 2: LC-MS/MS Data Acquisition
Phosphopeptides are analyzed on high-resolution Orbitrap platforms with optimized LC gradients. Stepped HCD fragmentation provides peptide identification with neutral loss signatures. EThcD is applied for unambiguous phosphorylation site localization. Multi-injection fractionation (high-pH RP, SAX, SCX) is used for deep phosphoproteome discovery projects.
Step 3: Phosphopeptide Identification and Site Localization
Raw MS data are searched using MaxQuant, Proteome Discoverer, and Philosopher with phosphorylation as a variable modification. Site localization is scored using MaxQuant localization probability, PhosphoRS, or Andromeda scores. Every identified phosphorylation site is validated against key quality criteria including localization probability ≥0.75, peptide backbone coverage, and spectral quality.
Step 4: Quantitative Analysis and Differential Expression
Phosphopeptide abundance is quantified using label-free (XIC alignment), TMT (reporter ion intensities), or SILAC (heavy/light ratios) approaches depending on experimental design. Differential expression analysis identifies significantly regulated phosphorylation sites across conditions with appropriate statistical testing and multiple hypothesis correction.
Step 5: Kinase-Substrate and Pathway Analysis
Phosphoproteomics data is integrated with kinase-substrate prediction databases (PhosphoSitePlus, KinBase, NetPhorest) to identify upstream kinases and regulated signaling networks. Motif analysis reveals enriched sequence preferences around regulated phosphorylation sites. Pathway enrichment analysis links phosphorylation changes to functional signaling cascades.
Step 6: Deliverables and Scientist Review
Complete phosphoproteomics dataset including phosphorylation site table with localization confidence scores, quantitative data with statistical analysis, annotated MS/MS spectra with site localization evidence, kinase-substrate network predictions, motif analysis results, pathway enrichment maps, and a scientist consultation session for biological interpretation.
For broader PTM discovery strategies beyond phosphorylation, our MS-Based PTM Analysis service provides a comprehensive framework for multi-modification discovery workflows.
Phosphoproteomics in Biomedical and Pharmaceutical Research
Phosphoproteomic analysis provides unique molecular insights across a broad spectrum of research areas. Our platform is configured to support the specific requirements of each application domain, from exploratory signaling studies in basic research to regulated characterization in drug development programs.
Cancer Signaling and Kinase Drug Discovery
Dysregulated kinase activity is a hallmark of cancer, and phosphoproteomics provides a direct readout of oncogenic signaling pathway activity. Our platform enables comprehensive profiling of phosphorylation changes in response to kinase inhibitor treatment, identification of drug resistance mechanisms through adaptive kinase reprogramming, and discovery of novel phosphorylation biomarkers for patient stratification. Phosphoproteomic analysis of tumor tissues and liquid biopsy samples reveals the functional state of signaling networks that cannot be inferred from genomic or transcriptomic data alone. Our kinase activity profiling platform provides complementary functional kinase activity measurements.
Cell Signaling and Systems Biology
Phosphorylation is the primary mechanism of rapid signal transduction in eukaryotic cells. Time-resolved phosphoproteomics captures the dynamic rewiring of signaling networks following growth factor stimulation, stress response, or cell cycle progression. Our workflow is optimized for multi-condition time-course experiments, enabling comprehensive mapping of phosphorylation cascades and feedback regulation. Integrated analysis of phosphoproteomics with proteomics and transcriptomics provides systems-level understanding of signaling network organization.
Neuroscience and Neurodegenerative Disease
Abnormal protein phosphorylation is a central feature of neurodegenerative diseases, including tau hyperphosphorylation in Alzheimer's disease and α-synuclein phosphorylation in Parkinson's disease. Our phosphoproteomics platform supports in-depth characterization of disease-relevant phosphorylation events in brain tissue, CSF, and cell models. For targeted analysis of specific neurodegenerative phosphorylation markers, our PRM PTM Verification service provides quantitative validation with high specificity and sensitivity.
For comprehensive quantitative analysis across large sample sets, our PTM Quantitative Analysis Services platform provides integrated multiplexed quantification workflows.
Case Study: Phosphoproteomics Reveals Signaling Transitions During Mosquito Blood Meal Processing
A 2022 study by Kandel et al. published in PLOS ONE applied exploratory phosphoproteomics profiling to investigate the dramatic functional transition of Aedes aegypti Malpighian tubules (MTs) during blood meal processing, demonstrating the power of global phosphoproteomic analysis to uncover signaling mechanisms underlying rapid physiological adaptation.
Background: Female mosquitoes undergo a rapid and massive physiological shift after blood feeding — within minutes, MT fluid secretion increases more than 1,000-fold to eliminate excess water and ions from the blood meal. This transition is too rapid to be explained by transcriptional changes alone, strongly implicating post-translational regulation, particularly protein phosphorylation, as the primary mechanism. However, the specific phosphorylation events and signaling pathways driving this functional transition were largely unknown.
Approach: The team performed global phosphoproteomic profiling of MTs dissected from non-blood-fed (NBF) and blood-fed (BF) Aedes aegypti using IMAC-based phosphopeptide enrichment followed by high-resolution LC-MS/MS. Phosphorylation sites were identified and quantified across conditions, with site localization confidence assessed using established scoring algorithms. Bioinformatic analysis including kinase-substrate prediction, motif enrichment, and pathway annotation was applied to identify activated signaling networks and upstream kinases.
Key Findings:
- A total of 1,440 unique phosphorylation sites were identified on 563 proteins from mosquito MTs, including sites on V-ATPase subunits, ion transporters, and cytoskeletal regulators — representing the first comprehensive phosphoproteomic dataset for this mosquito organ
- Blood meal feeding induced significant phosphorylation changes in 249 sites, with 137 sites upregulated and 112 downregulated, revealing a dramatic reprogramming of MT signaling networks following blood feeding
- Seven unique phosphorylation sites were identified within four V-ATPase subunits, suggesting that phosphorylation-mediated regulation of the V-ATPase proton pump is a key mechanism driving the rapid increase in fluid secretion after blood feeding
- Kinase-substrate prediction analysis identified several upstream kinases potentially responsible for the observed phosphorylation changes, providing testable hypotheses for future functional studies
- Phosphorylation of cytoskeletal and junctional proteins suggested coordinated regulation of MT cell architecture during the diuresis response
Significance: This study demonstrates that global phosphoproteomic profiling can uncover the signaling mechanisms underlying rapid physiological transitions that are inaccessible to transcriptomic or proteomic approaches alone. By identifying specific phosphorylation sites on V-ATPase subunits and ion transporters, the findings provide molecular targets for understanding and potentially disrupting mosquito diuresis. For researchers investigating phosphorylation signaling in any biological system, our phosphoproteomics platform provides the analytical depth to map phosphorylation events, identify regulated pathways, and generate mechanistic hypotheses.
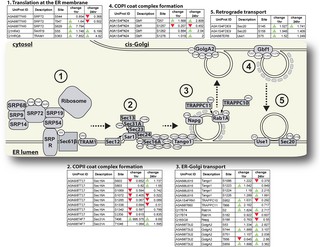
Figure 2 from Kandel et al. (2022). Phosphorylation mapping of Ae. aegypti Malpighian tubule proteins. (A) MT cell types: principal cells (PCs) and stellate cells (SCs). (B) V-ATPase-driven ion transport model. (C–D) Seven unique phosphorylation sites identified within four V-ATPase subunits, demonstrating phosphorylation-mediated regulation of the proton pump during blood meal-induced diuresis. (CC BY 4.0)
Representative Phosphoproteomics Data Outputs
Our phosphoproteomics pipeline delivers multi-dimensional data outputs that provide a complete picture of protein phosphorylation at site-specific resolution. Below are representative examples of the key data types included in every project deliverable. These outputs are generated for every identified phosphorylation site, enabling detailed assessment of phosphorylation identity, site localization confidence, and quantitative changes across conditions.
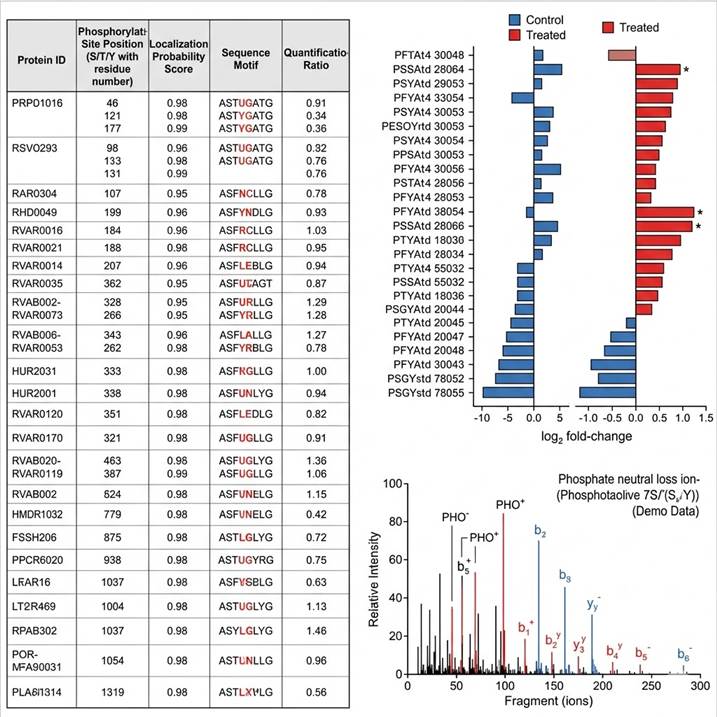
Representative phosphoproteomics data. (Left) Phosphorylation site identification table with protein ID, site position, localization probability, sequence motif, and ratios. (Center) Quantitative comparison bar charts showing phosphorylation level changes across conditions — significant sites highlighted in red. (Right) Annotated MS/MS spectrum with phosphate neutral loss ions and b/y-ion series confirming confident site localization.
Every data deliverable is reviewed by our phosphoproteomics scientists, who verify spectral quality, confirm site localization assignments, and provide biological context for the results. Custom visualization and data formatting options are available to match publication requirements or internal reporting standards.
Why Choose Our Phosphoproteomics Analysis Services
Optimized Enrichment and Fragmentation
Our platform combines multiple phosphopeptide enrichment strategies (IMAC, TiO₂, antibody-based) with multi-mode fragmentation (stepped HCD, EThcD) to maximize phosphoproteome coverage while maintaining confident site localization. Sequential enrichment strategies and multi-injection fractionation are deployed for deep phosphoproteome discovery projects requiring comprehensive coverage.
Multi-Platform Mass Spectrometry
Access to high-resolution Orbitrap and TIMS-TOF platforms allows us to match the instrumentation to the specific analytical requirements of each project. Nano-flow systems maximize sensitivity for limited samples, and ion mobility separation provides an additional dimension for resolving co-eluting phosphopeptides in complex samples.
Comprehensive Quantitative Strategies
We offer the full spectrum of quantification approaches — label-free, TMT, SILAC, and MRM/PRM — from discovery to targeted validation. Each strategy is optimized for the specific experimental design and biological question, ensuring appropriate statistical power and data quality.
Integrated Bioinformatics
Phosphoproteomics data interpretation requires specialized bioinformatics extending beyond standard identification workflows. Our analysis includes kinase-substrate prediction, motif enrichment, pathway annotation, and integration with public databases (PhosphoSitePlus, Phospho.ELM, NetPhorest, KinBase) for comprehensive biological context.
Related Services
Our phosphoproteomics analysis services are supported by a broader PTM characterization platform offering complementary analytical capabilities. These services can be combined to address multi-dimensional signaling network characterization needs.
Frequently Asked Questions
What is the difference between phosphoproteomics and traditional phosphorylation analysis?
Traditional phosphorylation analysis typically examines one or a few proteins at a time using phospho-specific antibodies or radioactive labeling. Phosphoproteomics uses mass spectrometry to simultaneously identify and quantify thousands of phosphorylation sites across the entire proteome from a single experiment. This global perspective enables unbiased discovery of regulated signaling networks, identification of novel phosphorylation events, and systems-level understanding of phosphorylation dynamics.
What enrichment methods do you use for phosphopeptides?
We deploy IMAC (Fe³⁺ or Ti⁴⁺) for broad phosphoproteome coverage, TiO₂/MOAC for complementary selectivity especially for multiply phosphorylated peptides, and antibody-based enrichment for targeted phosphotyrosine analysis. Sequential enrichment strategies combining IMAC and TiO₂ are used to maximize phosphoproteome depth. The enrichment strategy is selected based on sample type, amount, and research objective.
How do you ensure confident phosphorylation site localization?
We use EThcD fragmentation as the primary method for unambiguous site localization, as it preserves the phosphate group during peptide backbone fragmentation. Site localization confidence is scored using MaxQuant localization probability and PhosphoRS algorithms, with a threshold of ≥0.75 for high-confidence assignments. Ambiguous localizations are resolved through manual spectral inspection by experienced phosphoproteomics scientists.
What sample types are compatible with your phosphoproteomics workflow?
Our pipeline accepts a wide range of sample types including cultured cells (≥1×10⁷ cells), tissue samples (≥10 mg), biofluids including serum, plasma, and CSF (≥100 μL), and immunoprecipitated protein complexes. Phosphatase inhibitors must be included during sample collection and lysis to preserve endogenous phosphorylation. For limited samples, we offer optimized miniaturized protocols.
What quantification strategies are available for phosphoproteomics?
We offer label-free quantification using extracted ion chromatogram alignment for discovery studies, TMT labeling for multiplexed comparisons of up to 16 conditions, SILAC for metabolic labeling in cell culture models, and MRM/PRM methods with synthetic phosphopeptide standards for targeted absolute quantification. The choice depends on experimental design, sample type, and required precision.
Can you analyze tyrosine phosphorylation specifically?
Yes — tyrosine phosphorylation constitutes less than 1% of total cellular phosphorylation and requires specialized enrichment. We offer anti-phosphotyrosine antibody-based enrichment (using pan-pY antibodies such as pY100, 4G10, or PY20) combined with high-sensitivity LC-MS/MS to maximize coverage of this low-abundance but biologically critical modification. For detailed signaling pathway analysis, we combine pY enrichment with TMT quantification.
How does your bioinformatics analysis support phosphoproteomics data interpretation?
Our bioinformatics pipeline extends beyond standard identification workflows to include kinase-substrate prediction (using PhosphoSitePlus, KinBase, NetPhorest, and NetworkIN), phosphorylation motif analysis to identify enriched sequence contexts, pathway enrichment analysis to map regulated phosphorylation to functional signaling cascades, and integration with public phosphorylation databases for biological context. Results are delivered with interactive visualization and a scientist consultation session.
References
- Kandel Y, Pinch M, Lamsal M, Martinez N, Hansen IA. Exploratory phosphoproteomics profiling of Aedes aegypti Malpighian tubules during blood meal processing reveals dramatic transition in function. PLoS One. 2022;17(7):e0271248.
- Ochoa D, Jarnuczak AF, Viéitez C, Gehre M, Soucheray M, Mateus A, et al. The functional landscape of the human phosphoproteome. Nat Biotechnol. 2020;38(3):365-373.
- Lancaster NM, Sinitcyn P, Forny P, Peters-Clarke TM, Fecher C, Smith AJ, et al. Fast and deep phosphoproteome analysis with the Orbitrap Astral mass spectrometer. Nat Commun. 2024;15:7016.
For research use only. Not for use in diagnostic procedures.