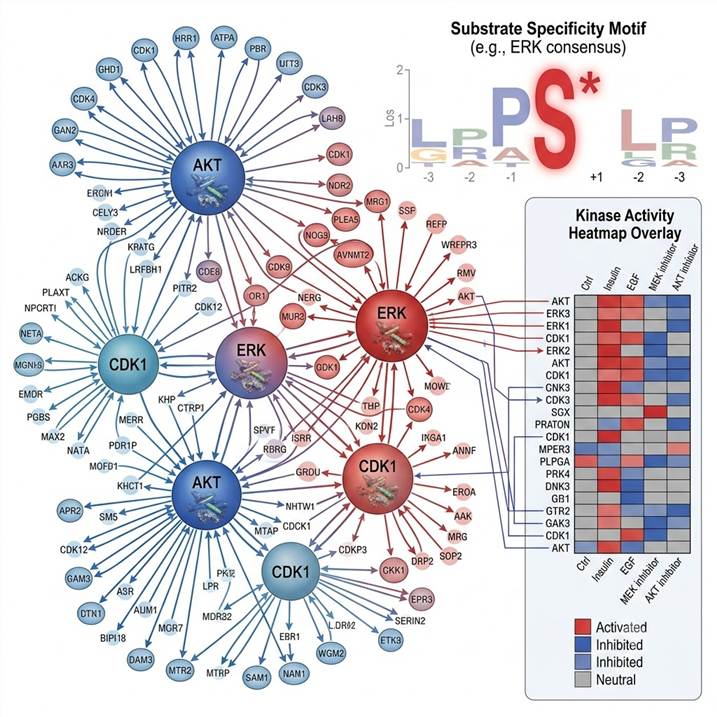
Why Kinase-Substrate Network Analysis Matters
A typical phosphoproteomics experiment yields thousands of regulated phosphorylation sites, but the functional interpretation of these sites is incomplete without answering two fundamental questions: which kinase is responsible? and what signaling network is being rewired?
Kinase-substrate network analysis transforms a list of phosphorylation sites into a mechanistic model of cellular signaling by:
- Identifying upstream kinases — Determining which kinases are activated or inhibited based on the collective phosphorylation patterns of their known and predicted substrates. This is especially valuable when kinase activity changes are not reflected at the protein expression level.
- Revealing signaling pathway crosstalk — Mapping connections between kinases and their substrates across canonical pathway boundaries reveals points of signal integration and divergence that are invisible when analyzing pathways in isolation.
- Prioritizing functional validation targets — Kinase-substrate predictions provide testable hypotheses for follow-up experiments. High-confidence predicted associations guide mutagenesis, inhibitor, and RNAi validation studies.
- Supporting drug mechanism-of-action studies — In drug development, understanding how a kinase inhibitor rewires the cellular signaling network (on-target and off-target effects) is critical for efficacy assessment and toxicity prediction. Our PTMs in Drug Discovery services integrate kinase network analysis with broader PTM drug development workflows.
- Enabling network-level biomarker discovery — Kinase activity signatures derived from substrate phosphorylation patterns provide more robust and reproducible biomarkers than individual phosphosite measurements.
Our Approach to Kinase-Substrate Network Inference
We deploy an integrated multi-algorithm pipeline that combines the strengths of several complementary approaches to achieve robust kinase-substrate network predictions.
Sequence-Based Kinase Prediction
Every phosphorylation site in your dataset is analyzed against known kinase consensus motifs. Using position-specific scoring matrices (PSSMs) derived from large-scale peptide array studies and curated phosphorylation databases, we score each site against the substrate specificity profiles of hundreds of human kinases. This approach captures the intrinsic sequence determinants of kinase-substrate recognition and provides predictions even for kinases with few known substrates.
Network-Based Inference with Functional Context
Sequence-based prediction alone misses the important biological context provided by protein-protein interactions, co-expression relationships, and shared pathway membership. Our pipeline integrates multiple network layers — kinase-kinase association networks and phosphosite-phosphosite association networks — to propagate information from well-characterized kinase-substrate pairs to understudied kinases and orphan phosphosites. This network-based approach significantly improves prediction coverage, particularly for the large fraction of phosphorylation sites with unknown upstream kinases.
Kinase Activity Inference from Substrate Phosphorylation Patterns
Rather than treating each phosphosite independently, our kinase activity inference module evaluates the collective phosphorylation behavior of all known and predicted substrates for each kinase. A kinase is inferred to be active when its substrate set shows coordinated regulation (enrichment of up- or down-regulated phosphosites). This approach detects kinase activity changes even when the kinase itself is not identified in the phosphoproteomics dataset — a common scenario because many kinases are expressed at low levels or are not detected by standard LC-MS workflows.
Integrated Database Resources
Our analysis draws on multiple curated and community resources to maximize prediction accuracy and coverage:
- PhosphoSitePlus — Curated phosphorylation sites and kinase-substrate relationships
- Kinase-substrate prediction databases — NetKSA, NetworkIN, and sequence-based motif libraries
- Pathway databases — KEGG, Reactome, and Gene Ontology for functional enrichment context
- Protein-protein interaction networks — STRING and BioGRID for network propagation scaffolds
The resulting kinase-substrate networks are cross-referenced against known biology and scored with confidence metrics, enabling you to focus on the most robust predictions for experimental follow-up. For broader PTM site annotation beyond kinase predictions, our PTM Site Prediction service provides additional computational tools for modification site discovery and characterization.
Compatible Data Types and Requirements
Our kinase-substrate network analysis pipeline accepts data from virtually any phosphoproteomics workflow. Sample requirements vary by project scope and analytical depth.
Input Data Formats
| Data Type |
Description |
Required Fields |
| Phosphosite identification list |
List of identified phosphosites from any DDA, DIA, or targeted phosphoproteomics pipeline |
Protein ID, phosphosite position (e.g., Ser473), sequence window (±7 amino acids), confidence score |
| Quantitative phosphoproteomics |
Label-free, SILAC, TMT/iTRAQ, or other quantitative comparisons between conditions |
All of the above + fold-change, p-value or significance score, intensity values |
| Time-course phosphoproteomics |
Multi-time-point datasets for dynamic signaling network reconstruction |
All of the above + time-point labels, temporal profiling patterns |
| Perturbation phosphoproteomics |
Kinase inhibitor, siRNA/CRISPR, or activator treatment datasets |
All of the above + perturbation conditions, dose-response metrics where applicable |
Recommended Project Scale
- Standard project: 500–5,000 quantified phosphosites, 1–10 conditions, ≥2 biological replicates per condition
- Deep discovery project: 5,000–30,000+ phosphosites, multi-condition or time-course designs with ≥3 replicates
- Targeted validation: 10–100 selected phosphosites with known or candidate upstream kinases
For comprehensive kinase-substrate analysis integrated with broader PTM discovery, our Phosphoproteomics Data Analysis service provides end-to-end bioinformatics support from raw data processing through network interpretation.
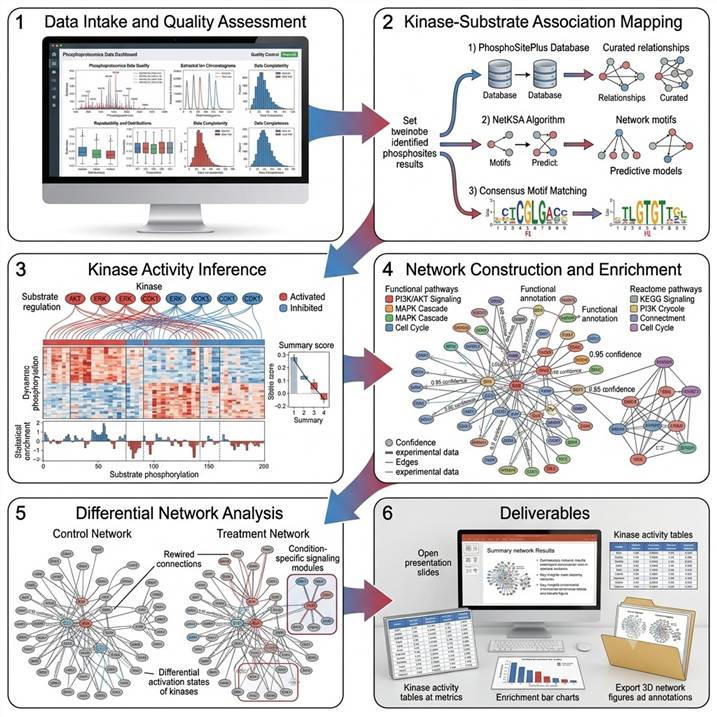
Workflow: From Phosphoproteomics Data to Signaling Network Models
Step 1: Data Intake and Quality Assessment
We review your phosphoproteomics dataset for data quality, coverage, and experimental design. Data from any platform (Orbitrap, TIMS-TOF, Q-TOF) and any quantification strategy (label-free, SILAC, TMT) is supported. Key quality metrics — phosphosite localization confidence, replicate reproducibility, and data completeness — are documented before analysis proceeds.
Step 2: Kinase-Substrate Association Mapping
Each phosphorylation site in your dataset is mapped against multiple kinase-substrate association resources: known kinase-substrate relationships from curated databases (PhosphoSitePlus), sequence-based kinase consensus motif predictions, and network-based kinase-substrate associations via NetKSA and complementary algorithms. Multiple prediction sources are integrated to improve coverage and confidence.
Step 3: Kinase Activity Inference
Kinase activity is inferred by evaluating the collective phosphorylation status of each kinase's substrate set. A kinase is scored as activated when its substrates show significant enrichment of up-regulated phosphosites, and inhibited when its substrates show enrichment of down-regulated sites. Permutation-based statistics provide significance estimates for each activity call.
Step 4: Network Construction and Enrichment
Predicted and known kinase-substrate relationships are assembled into a signaling network graph. Network nodes represent kinases and substrates; edges represent supported associations. The network is enriched with pathway annotations (KEGG, Reactome), functional categories (GO), and protein interaction context (STRING), enabling multi-layered biological interpretation.
Step 5: Differential Network Analysis
For multi-condition experiments, we perform comparative network analysis to identify condition-specific signaling modules, rewired kinase-substrate relationships, and differentially activated kinase networks. Topological analysis identifies network hubs, bottlenecks, and signaling nodes that bridge multiple pathways.
Step 6: Deliverables and Review
Publication-ready kinase-substrate network figures (2D and 3D), kinase activity heatmaps and enrichment tables, full kinase-substrate association tables with confidence scores, network topology analysis report, functional enrichment results, and a scientist consultation session for biological interpretation.
Why Choose Our Kinase-Substrate Network Analysis Service
Multi-Algorithm Integration for Maximum Coverage
No single kinase prediction algorithm achieves complete coverage. Our pipeline integrates sequence-based, network-based, and database-driven approaches, ensuring that both well-studied and understudied kinases are represented in your network models.
Publication-Ready Visual Outputs
Network figures are formatted to publication standards from the outset — including proper color schemes, legible node labels, pathway annotations, and multiple export formats (SVG, PDF, TIFF). Our visualizations are designed to be presentation- and manuscript-ready without additional editing.
Expert Biological Interpretation
Our team includes scientists with deep domain expertise in kinase signaling, phosphoproteomics, and computational biology. Every project includes a consultative data review session where we walk through the network results, highlight the most robust findings, and discuss experimental follow-up strategies.
Flexible Analytical Depth
Whether you need a targeted analysis of 50 phosphosites around a specific signaling pathway or a systems-level reconstruction of the entire kinome-substrate network from a deep phosphoproteomics dataset, our pipeline scales to match your research question. For broader integration of PTM datasets, our PTM Bioinformatics Analysis service provides multi-omics functional interpretation and pathway mapping.
Case Study: NetKSA — Network-Based Kinase-Substrate Association Prediction Using Functional Landscapes
In a 2023 study published in Pacific Symposium on Biocomputing, Ayati et al. developed NetKSA, a network-based machine learning framework that substantially improves kinase-substrate association prediction by integrating heterogeneous functional data.
Background: Over 90% of identified phosphorylation sites have no known upstream kinase. The vast majority of known kinase-substrate associations are concentrated on only ~20% of well-studied kinases, leaving the signaling networks controlled by less-characterized kinases largely unexplored. Existing prediction methods rely primarily on local sequence motifs and miss the broader functional context.
Approach: NetKSA constructs two complementary networks: a phosphosite-phosphosite association network (integrating sequence similarity, shared biological pathways, co-evolution, and co-phosphorylation across biological states) and a kinase-kinase association network (integrating protein-protein interactions, shared pathways, and kinase family membership). Node embeddings from these heterogeneous networks are used as feature vectors to train machine learning models for kinase-substrate association prediction. The framework is robust to the choice of network embedding algorithm, and each network type provides complementary information.
Key Findings:
- NetKSA outperforms two state-of-the-art algorithms — KinomeXplorer and LinkPhinder — in overall kinase-substrate association prediction accuracy
- The kinase-kinase and phosphosite-phosphosite network layers provide complementary predictive power; combining both networks outperforms either alone
- NetKSA enables annotation of phosphosites targeted by less-studied kinases through a kinase stratification strategy based on known substrate counts
- The network-based approach captures contextual information — including pathway relationships and co-regulation across conditions — that sequence-only methods cannot access
- Performance is robust across different network embedding algorithms, demonstrating generalizability of the network integration strategy
Significance: This study demonstrates that network-based integration of functional genomic and phosphoproteomic data substantially improves kinase-substrate association prediction compared to sequence-based approaches alone. The framework provides a pathway to systematically annotate orphan phosphosites and expand coverage of understudied kinases — both critical bottlenecks in phosphoproteomics data interpretation.
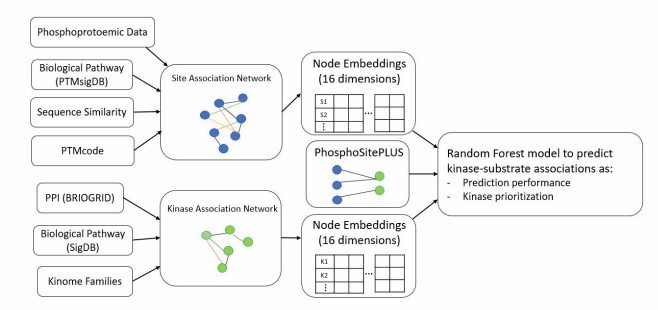
Figure 1 from Ayati et al. (2023). NetKSA workflow: heterogeneous network construction, node embedding, and machine learning-based kinase-substrate association prediction. (CC BY 4.0)
Representative Kinase-Substrate Network Analysis Results
Our kinase-substrate network analysis delivers integrated data packages with multiple visualization and annotation layers, enabling immediate biological interpretation and publication-quality figure generation.
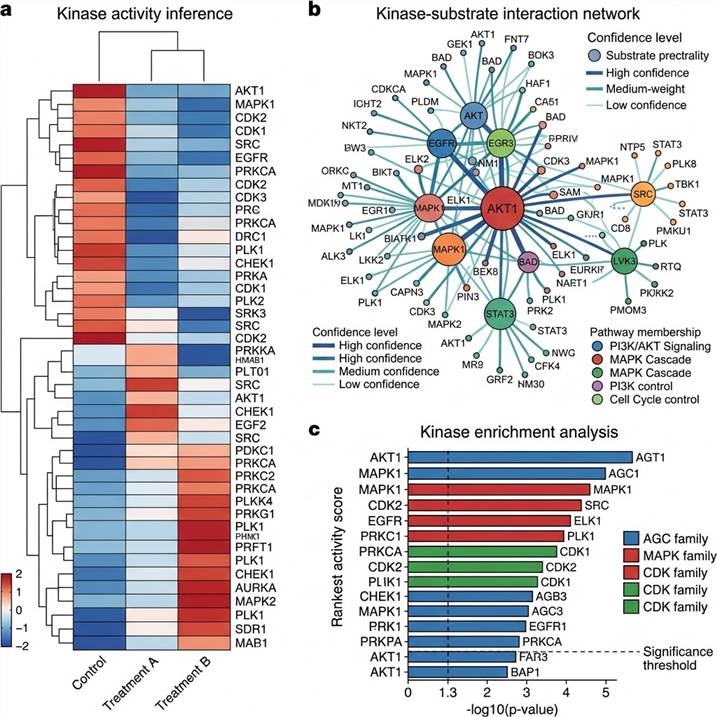
Representative data outputs from our kinase-substrate network analysis pipeline. Left: Kinase activity inference heatmap across experimental conditions. Center: Kinase-substrate interaction network with pathway annotations. Right: Kinase activity enrichment analysis with functional context.
Key deliverables included in every project package:
- Kinase activity heatmap — Inferred activation or inhibition scores for all detectable kinases across experimental conditions, with statistical significance
- Kinase-substrate interaction network — Graph visualization with nodes (kinases and substrates), edges (known and predicted associations), pathway annotations, and confidence scoring
- Kinase enrichment table — For each kinase, the list of regulated substrate phosphosites with fold-changes, p-values, and prediction confidence scores
- Network topology report — Identification of network hubs, bottleneck nodes, and condition-specific signaling modules
- Pathway enrichment — KEGG and Reactome pathway enrichment analysis for kinasetargeted and regulated substrate sets
Related Services
Our kinase-substrate network analysis is one component of a comprehensive phosphoproteomics and bioinformatics platform. These services can be used independently or integrated into a complete signaling analysis workflow.
FAQs
What is kinase-substrate network analysis?
Kinase-substrate network analysis is a bioinformatics approach that maps phosphorylation sites in your dataset to the kinases that are likely responsible for phosphorylating them. By integrating sequence motifs, protein-protein interactions, pathway context, and curated kinase-substrate databases, we reconstruct the upstream signaling networks that drive the observed phosphorylation landscape.
What type of data do I need to provide?
We accept phosphoproteomics data from any platform and quantification strategy. The minimum requirement is a list of identified phosphosites with protein identifiers, site positions, and sequence windows (±7 amino acids). For differential network analysis, quantitative data with fold-changes and significance values across conditions is preferred.
Can you predict kinases for non-human species?
Yes — for model organisms with well-characterized kinomes (mouse, rat, zebrafish, Drosophila, C. elegans, Arabidopsis, yeast), we can perform kinase-substrate mapping using species-specific kinase databases and orthology-based predictions. Coverage depends on the completeness of kinase-substrate annotations for the target species.
How accurate are kinase-substrate predictions?
Prediction accuracy varies by kinase and substrate type. For well-studied kinases with defined consensus motifs, prediction precision is high. For understudied kinases, predictions are less certain but provide valuable hypotheses for experimental validation. We provide confidence scores for every predicted association and recommend orthogonal validation for critical targets.
What kind of network visualizations do you provide?
We provide publication-ready 2D network figures with pathway annotations, as well as interactive network files compatible with Cytoscape and Gephi for further exploration. Visualization options include kinase-centered views, substrate-centered views, pathway-focused subnetworks, and comparative networks for multi-condition experiments.
Can you analyze data from kinase inhibitor experiments?
Yes — perturbation phosphoproteomics is one of our core application areas. Kinase inhibitor datasets are analyzed to identify on-target and off-target effects, reconstruct drug-rewired signaling networks, and infer inhibitor mechanism of action from substrate phosphorylation patterns.
How long does a typical project take?
A standard project (500–5,000 phosphosites, ≤5 conditions) typically completes in 2–3 weeks from data receipt to final deliverable. Larger projects (10,000+ phosphosites, multi-condition or time-course) require 3–5 weeks. Timelines are confirmed during the initial project consultation.
What if my phosphoproteomics data was generated by a different service provider?
No problem. Our kinase-substrate network analysis can be performed on data from any source. We accept standard output formats from MaxQuant, Proteome Discoverer, FragPipe, Spectronaut, and other common phosphoproteomics pipelines. If needed, we can assist with data format conversion.
References
- Ayati M, Yilmaz S, Lopes FBT, Chance M, Koyuturk M. Prediction of Kinase-Substrate Associations Using The Functional Landscape of Kinases and Phosphorylation Sites. Pac Symp Biocomput. 2023;28:73-84.
- Franciosa G, Locard-Paulet M, Jensen LJ, Olsen JV. Recent advances in kinase signaling network profiling by mass spectrometry. Curr Opin Chem Biol. 2023;73:102260.
- Anandakrishnan M, Ross KE, Chen C, Shanker V, Cowart J, Wu CH. KSFinder — a knowledge graph model for link prediction of novel phosphorylated substrates of kinases. PeerJ. 2023;11:e16164.
For research use only. Not for use in diagnostic procedures.