Why Succinylome Profiling Matters Now — A Metabolic Sensor with Drug Target Potential
Succinylation differs from most PTMs in one critical respect: it has a genuine non-enzymatic component that scales with metabolite concentration. In mitochondria, where succinyl-CoA concentrations range from 20 to 200+ µM depending on metabolic state, the probability of spontaneous lysine succinylation is physically significant. This means the succinylome is not merely a catalog of enzyme-substrate relationships — it is a real-time chemical footprint of TCA cycle activity.
This property transforms succinylome data from descriptive to diagnostic. When a tumor harbors a succinate dehydrogenase (SDH) mutation, succinate accumulates to millimolar levels, driving succinyl-CoA abundance upward and producing a hyper-succinylation phenotype that remodels the mitochondrial proteome. In isocitrate dehydrogenase (IDH)-mutant glioma, the 2-hydroxyglutarate (2-HG) oncometabolite alters succinyl-CoA metabolism indirectly. When fumarate hydratase (FH) is lost in renal cancer, fumarate accumulation triggers succinylation of KEAP1 at critical cysteine residues, activating NRF2-mediated antioxidant programs. In each case, succinylome profiling reveals the connection between a genetic lesion and its proteomic consequences — information that genomics alone cannot provide.
For drug discovery groups, this is actionable. The demonstration that STAT1 succinylation at K410 and K413 creates a druggable node in silicosis-associated lung fibrosis (see Case Study) illustrates a general principle: PTM-regulated transcription factors and metabolic enzymes are increasingly recognized as therapeutic targets, and succinylome data is the necessary starting point for identifying which modification events are functionally relevant.
The convergence of three trends makes now the right time for succinylome investment:
- Improved enrichment reagents: Motif-specific anti-succinyllysine antibodies now achieve enrichment efficiencies that enable detection of low-stoichiometry (1–10%) modification events that were invisible five years ago.
- Multiplexed quantification: TMT-based workflows (6-plex to 16-plex) enable statistically powered comparisons across conditions, time points, and genetic backgrounds in a single experiment.
- Bioinformatics maturity: SIRT5 substrate predictors (GPSuc), succinylation-specific motif analysis tools (Motif-x), and integrated PTM databases (PhosphoSitePlus, dbPTM) now provide the analytical infrastructure to extract mechanistic insight from succinylome data rather than just producing site lists.
The SIRT5 Regulatory Axis — Enzyme-Directed vs. Non-Enzymatic Succinylation in Disease
SIRT5 is the dominant desuccinylase in mammalian cells, but its biology is nuanced in ways that directly affect experimental design for succinylome studies.
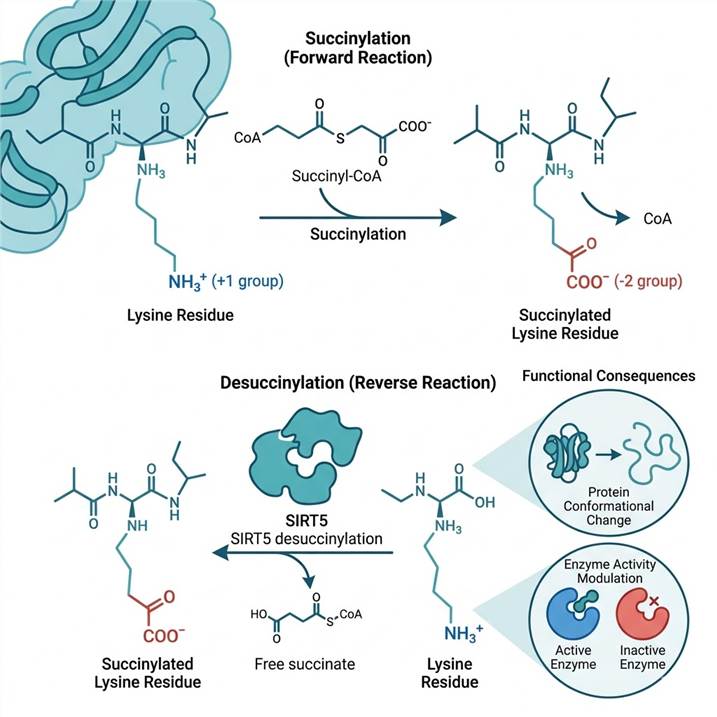
SIRT5 has three catalytic activities — desuccinylation, demalonylation, and deglutarylation — but negligible deacetylase activity. Structural studies reveal that SIRT5's substrate-binding pocket contains an arginine residue (Arg105) and a tyrosine residue (Tyr102) that form ionic and hydrogen-bonding interactions with the carboxylate group of succinyl/malonyl/glutaryl-lysine, interactions that cannot form with the methyl group of acetyl-lysine. This explains the ~1,000× selectivity for succinyl over acetyl substrates and makes SIRT5 a functionally dedicated short-chain acyl-lysine eraser.
SIRT5 knockout phenotypes are conditional. SIRT5−/− mice are viable and fertile with no overt developmental abnormalities, but when challenged with high-fat diet they develop hepatic steatosis, impaired fatty acid oxidation, and reduced ketogenesis — phenotypes traceable to hyper-succinylation of rate-limiting β-oxidation enzymes (HADHA at K351, ACADM at multiple sites). Under fasting conditions, SIRT5−/− mice fail to upregulate ketone body production because HMGCS2, the mitochondrial rate-limiting enzyme of ketogenesis, requires SIRT5-mediated desuccinylation at K83 and K310 for full activity. Under cold exposure, brown adipose tissue from SIRT5−/− mice shows impaired thermogenesis linked to hyper-succinylation of UCP1.
In cancer, SIRT5 plays context-dependent roles. In hepatocellular carcinoma, SIRT5 suppresses tumor growth by desuccinylating and inactivating citrate synthase (CS) at K375, reducing mitochondrial respiration and promoting apoptosis. In chordoma, SIRT5 is oncogenic — it desuccinylates c-myc at K369, stabilizing the oncoprotein and driving proliferation. These opposing roles mean that succinylome data must be interpreted in tissue- and disease-specific context, not according to a universal “more succinylation = good/bad” rule.
The non-enzymatic problem is real. At succinyl-CoA concentrations above 50 µM — which occur physiologically in mitochondria during active TCA cycle flux, and pathologically in SDH-deficient tumors — non-enzymatic succinylation contributes substantially to the total succinylome. The SIRT5-regulon classification approach addresses this: sites whose succinylation levels change significantly upon SIRT5 knockdown/knockout or pharmacological SIRT5 inhibition are classified as enzyme-regulated; sites that are insensitive to SIRT5 perturbation are candidate non-enzymatic modifications. This stratification is included in our standard bioinformatics pipeline.
Our Succinylation Proteomics Platform — Anti-Succinyllysine Enrichment to High-Resolution LC-MS/MS
Succinylation analysis presents two analytical challenges: the PTM is low stoichiometry (1–10% of a given lysine pool) and chemically indistinguishable from other acyl modifications at the MS1 level without enrichment. Our five-step workflow addresses both with pre-fractionation for dynamic range, motif-specific antibody enrichment, and high-resolution Orbitrap MS.
Step 1: Protein Extraction & Digestion
Samples are lysed under denaturing conditions (8 M urea) with deacylase inhibitors (nicotinamide, trichostatin A) to prevent in vitro deacylation. Proteins are reduced, alkylated, and digested with sequencing-grade trypsin (1:50 ratio, 37°C, overnight).
Step 2: High-pH RP Fractionation
Tryptic peptides are separated into 8–12 fractions by high-pH RP-HPLC before enrichment. This is critical: succinylated peptides are 10–100× lower in abundance than unmodified species, and fractionation prevents ion suppression from masking them during LC-MS/MS.
Step 3: Anti-Succinyllysine Enrichment
Each fraction undergoes immunoaffinity enrichment with anti-succinyllysine antibody-conjugated beads (PTMScan® grade). The antibody recognizes the succinyl-K epitope with minimal cross-reactivity toward acetyl-, malonyl-, or glutaryl-lysine. Enriched peptides are eluted with 0.15% TFA and desalted on C18 StageTips.
Step 4: nano-LC-MS/MS (Orbitrap)
Enriched peptides are separated on a 50 cm × 75 µm nano-LC column (120-min gradient) and analyzed on a Q Exactive HF-X or Exploris 480. Full MS at 120,000 resolution; top-20 HCD fragmentation at 28% NCE; MS/MS at 15,000 resolution.
Step 5: Data Analysis & QC
Raw files are searched with MaxQuant (or Proteome Discoverer/Sequest HT). Variable modifications: succinylation (K, +100.01604 Da), acetylation (K, +42.01056 Da), oxidation (M). FDR <1% at PSM/peptide/protein levels. Site localization probability >0.75 required for reporting.
QC benchmarks reported per project: total succinylation sites identified, sites with localization probability >0.75, enrichment specificity (% of identified peptides carrying succinyl-K), and for quantitative projects, the correlation coefficient (R²) between biological replicates.
Quantitative Succinylome Profiling & Site Occupancy Analysis
Succinylation is intrinsically quantitative biology — the magnitude of modification at a given site matters as much as its presence or absence. We offer two complementary quantification strategies and a unique site occupancy module that no competitor currently provides.
TMT Multiplexed Quantification
Recommended for defined comparisons (WT vs. KO, treated vs. control, tumor vs. normal). TMTpro 6-plex to 16-plex labeling with SPS-MS3 quantification on Orbitrap delivers accurate relative quantification with minimal ratio compression. The Case Study below used this approach to quantify 4,588 succinylation sites across silicosis and control tissue in a single TMT 6-plex experiment.
Label-Free Quantification (LFQ)
Suitable for larger cohorts, clinical specimen sets, or projects exceeding TMT capacity. LFQ uses MS1 peak area integration with total ion current normalization and retention time alignment. Recommended for >16 samples or when sample amount is limiting.
Site Occupancy (Stoichiometry) Analysis
Unique differentiator — zero competitors offer this. Using parallel digestion (one aliquot for succinyl-peptide enrichment, the other for total proteome), MS1-based ratio calculations estimate what fraction of each lysine pool is succinylated. Example output: “CS K375 is 8.3% succinylated in tumor vs. 2.1% in normal” — providing the stoichiometric context to judge functional significance.
Expected depth by sample type:
| Sample Type |
Typical Succinylation Sites Identified |
Proteins Covered |
| Mammalian cell line (e.g., HEK293T, HeLa) |
3,000 – 5,000 |
1,500 – 2,500 |
| Mouse/human tissue (liver, heart, brain) |
2,000 – 4,000 |
1,200 – 2,000 |
| Mitochondrial-enriched fraction |
800 – 1,500 |
400 – 700 |
| Plant tissue |
1,500 – 3,000 |
800 – 1,500 |
Multi-Acyl Crosstalk Profiling — Succinylation in the PTM Network
Succinylation does not occur in isolation. The mitochondrial acyl-CoA pool includes acetyl-CoA, succinyl-CoA, malonyl-CoA, glutaryl-CoA, and several other short-chain acyl-CoA species — all of which can serve as acyl donors for lysine modification. The TCA cycle sits at the hub of this network: citrate → acetyl-CoA for acetylation, α-ketoglutarate → succinyl-CoA for succinylation, and malonyl-CoA (derived from acetyl-CoA carboxylase) for malonylation.
Sequential enrichment from a single digest enables co-profiling of multiple acyl-PTM types from the same sample. The workflow uses successive rounds of immunoaffinity enrichment: first anti-acetyllysine beads, then anti-succinyllysine beads, applied to the same pre-fractionated peptide pool. For projects requiring malonylome data, a third anti-malonyllysine enrichment is added. Each enriched fraction is analyzed by separate LC-MS/MS runs.
Crosstalk analysis deliverables include:
- Site competition maps: Identification of lysine residues that are modified by both succinylation and acetylation — these are sites where the two modifications compete for the same residue and likely act as a regulatory switch.
- Mutual exclusivity analysis: Statistical assessment of whether succinylation and acetylation at the same site are correlated, anti-correlated, or independent across conditions.
- Pathway-level integration: KEGG pathway maps annotated with succinylation, acetylation, and expression-level data, revealing how different PTM layers converge on the same biological processes.
The multi-acyl approach is particularly informative for studies of SIRT5 function, since SIRT5 demalonylates and deglutarylates in addition to desuccinylating — all three activities are lost upon SIRT5 deletion. Co-profiling succinylation, malonylation, and glutarylation provides a complete picture of the SIRT5-regulated acylome.
Applications in Cancer Metabolism — Mapping Succinylome Rewiring in Tumors
Cancer succinylome research has coalesced around three genetic-metabolic contexts where succinyl-CoA homeostasis is disrupted:
SDH-deficient tumors (GIST, pheochromocytoma, paraganglioma). Loss-of-function mutations in succinate dehydrogenase subunits (SDHA/B/C/D) cause succinate accumulation to millimolar concentrations. This metabolic crisis has two succinylation-related consequences: first, the massive succinyl-CoA pool drives non-enzymatic hyper-succinylation of mitochondrial proteins; second, succinate competitively inhibits α-ketoglutarate-dependent dioxygenases (TET, JMJD histone demethylases), creating an epigenetic phenotype that synergizes with succinylome remodeling. Quantitative succinylome comparison of SDH-mutant vs. SDH-wild-type tumors reveals which metabolic enzymes are functionally impacted by hyper-succinylation and identifies potential collateral vulnerabilities.
IDH-mutant glioma and AML. IDH1/2 mutations produce the oncometabolite 2-hydroxyglutarate, which does not directly succinylate proteins but rewires mitochondrial metabolism in ways that alter succinyl-CoA availability. Global succinylome profiling in IDH-mutant cells has identified succinylation changes on proteins in the TCA cycle itself (IDH2, OGDH) and on chromatin regulators, suggesting a feed-forward mechanism connecting oncometabolite production to PTM-mediated metabolic and epigenetic dysregulation.
SIRT5 in hepatocellular carcinoma. Citrate synthase (CS) is hyper-succinylated at K375 in HCC tumor tissue compared to adjacent normal liver. SIRT5-mediated desuccinylation of CS at this site inactivates the enzyme, reducing mitochondrial respiration and ATP production — a tumor-suppressive mechanism. Conversely, in the same tumor type, KAT2A acts as a succinyltransferase, succinylating the splicing factor SRSF11 to promote HCC progression. These opposing activities illustrate why context-specific succinylome profiling, not just SIRT5 expression measurement, is needed to interpret succinylation biology in cancer.
Beyond metabolism — succinylation of non-metabolic proteins. While the mitochondrial succinylome dominates in terms of site numbers, nuclear and cytosolic succinylation events are increasingly recognized as functionally important. Histone succinylation at H3K122, catalyzed by SIRT7, regulates chromatin compaction. Transcription factor succinylation — STAT1 (silicosis), c-myc (chordoma), p53 — connects metabolic state directly to gene expression programs.
Applications in Metabolic Disease & Mitochondrial Dysfunction
NAFLD/NASH. The progression from simple steatosis to steatohepatitis involves a metabolic transition where mitochondrial TCA cycle flux becomes dysregulated, leading to succinyl-CoA accumulation. SIRT5 expression is downregulated in fatty liver, reducing desuccinylation capacity and shifting the succinylome toward hyper-modification of fatty acid β-oxidation enzymes. Succinylome profiling in mouse NASH models has identified hyper-succinylation of HADHA (K351, K560), ACADM (K243), and ECHS1 — all core β-oxidation components — as a molecular signature of disease progression. These sites are candidate biomarkers for distinguishing simple steatosis from NASH.
Diabetes and pancreatic β-cell dysfunction. Mitochondrial succinylation in pancreatic β-cells is sensitive to glucose concentration through succinyl-CoA production rates. SUCLA2, the β-subunit of succinyl-CoA synthetase, is itself regulated by SIRT5-mediated desuccinylation — creating an autoregulatory loop where the enzyme that produces succinyl-CoA is feedback-inhibited by the succinylation it generates, unless SIRT5 removes the modification. Disruption of this loop in diabetic models contributes to mitochondrial dysfunction and impaired glucose-stimulated insulin secretion.
Cardiac metabolism. The heart is the most mitochondrion-dense tissue in the body and correspondingly has a rich succinylome. In diabetic cardiomyopathy, Sirt5 expression is modestly elevated as a compensatory response to lipid overload, and Sirt5 overexpression in cardiomyocytes improves fatty acid metabolism and reduces lipotoxicity. Succinylome profiling of cardiac tissue from heart failure models has identified succinylation changes on SDHA and ATP5A (ATP synthase subunit α), linking metabolic sensing directly to the electron transport chain and ATP production machinery.
Neurodegeneration. Tau protein succinylation at K311 has been mechanistically linked to Alzheimer's disease pathology: succinylation at this site promotes tau aggregation and reduces microtubule binding, and succinylated tau is enriched in AD brain tissue compared to age-matched controls. Amyloid precursor protein (APP) is also succinylated, with modification status affecting its proteolytic processing. Mitochondrial protein succinylation is globally altered in AD brain, suggesting that TCA cycle-derived succinyl-CoA dysregulation contributes to the metabolic component of neurodegeneration.
Case Study — Integrated Succinylome, Acetylome & Proteome Profiling Reveals STAT1 as a Drug Target in Fibrotic Disease
Reference: Zhang T, Wang Y, Sun Y, et al. Proteome, Lysine Acetylome, and Succinylome Identify Posttranslational Modification of STAT1 as a Novel Drug Target in Silicosis. Molecular & Cellular Proteomics. 2024;23(6):100770. DOI: 10.1016/j.mcpro.2024.100770 (CC BY 4.0)
The Problem. Silicosis is a progressive, irreversible fibrotic lung disease caused by inhalation of crystalline silica particles. Despite decades of research, no pharmacological therapy halts or reverses silicosis progression. The molecular mechanisms linking silica exposure to macrophage activation, inflammation, and fibrosis remain incompletely understood — and no prior study had systematically examined PTM-level changes in silicotic lung tissue.
The Approach. Zhang et al. applied a multi-omics strategy to a mouse silicosis model. Lung tissue from silica-exposed C57BL/6J mice was compared to saline-instilled controls using TMT 6-plex labeling. Three data layers were acquired from the same tissue: (1) total proteome — quantitative comparison of protein abundance; (2) acetylome — anti-acetyllysine enrichment + LC-MS/MS; (3) succinylome — anti-succinyllysine enrichment + LC-MS/MS. Peptides were pre-fractionated by high-pH RP-HPLC (8 fractions), enriched with motif-specific antibodies, and analyzed on a Q Exactive Plus mass spectrometer. Data were processed with MaxQuant (FDR < 1%) and differential analysis was performed with Benjamini-Hochberg correction.
Key Results. The succinylome analysis identified 4,588 unique succinylation sites on 2,263 proteins, of which 4,032 sites passed QC for quantitative comparison. This represents one of the deepest mammalian succinylomes reported to date. The multi-omics integration revealed that transcription factor STAT1 was simultaneously regulated at the expression, acetylation, and succinylation levels in silicotic tissue: STAT1 protein abundance increased 2.3-fold; STAT1 acetylation at K410 and K413 increased significantly; STAT1 succinylation at the same K410/K413 residues was also elevated. Functional experiments demonstrated that STAT1 K410/K413 PTM status regulated its transcriptional activity and nuclear translocation. Pharmacological intervention with geranylgeranylacetone (GGA) — an acyclic polyisoprenoid with anti-inflammatory properties — reversed STAT1 hyper-modification and attenuated silica-induced pulmonary fibrosis in the mouse model.
Why This Matters for Your Project. This study exemplifies the translational value of integrated PTM proteomics. Without the succinylome layer, STAT1 would have been identified as an upregulated protein — interesting but insufficient for drug targeting. The acetylation and succinylation data at single-residue resolution (K410, K413) provided the mechanistic specificity needed to nominate STAT1 as a target and GGA as a repositioning candidate. This is the type of outcome that integrated proteome + acylome analysis can deliver for oncology, fibrosis, and metabolic disease projects.
Sample Requirements, Bioinformatics & Data Deliverables
Sample Requirements
| Sample Type |
Recommended Amount |
Minimum Amount |
| Cultured cells (mammalian) |
1 × 107 cells |
5 × 106 cells |
| Fresh-frozen tissue |
50 mg |
20 mg |
| Mitochondrial-enriched fraction |
200 µg protein |
100 µg protein |
| FFPE tissue |
10 sections (10 µm, 100 mm²) |
5 sections |
Biological replicates: ≥3 per condition for quantitative comparison. For animal studies, we recommend 4–6 animals per group. For clinical specimens, 6–10 samples per cohort.
Sample preparation guidelines: Flash-freeze tissue in liquid nitrogen within 10 minutes of collection. Add deacylase inhibitors (nicotinamide 5 mM + trichostatin A 1 µM) to lysis buffer if preparing lysates yourself. Ship on dry ice.
Bioinformatics Deliverables
Every succinylome project includes a standardized analysis package:
Core quantitative outputs:
- Succinylation site table (UniProt accession, gene symbol, peptide sequence, Ksu position, localization probability, fold change, p-value, adjusted p-value per comparison)
- Motif-x sequence motif analysis (±10 amino acids around succinylated lysine)
- SIRT5 substrate prediction scores (GPSuc 2.0 database cross-reference)
- Subcellular localization annotation (Gene Ontology Cellular Component)
- Volcano plots and hierarchical clustering heatmaps (per comparison)
- Principal component analysis (PCA) for sample grouping validation
Pathway and network analysis:
- KEGG pathway enrichment (hypergeometric test, Benjamini-Hochberg FDR correction)
- Gene Ontology (Biological Process, Molecular Function) enrichment
- Protein-protein interaction network (STRING database, confidence score >0.7)
- Kinase-substrate relationship mapping (if co-profiled with phosphoproteomics)
Publication-ready figures and resources:
- Ready-to-edit Methods section text for manuscript preparation
- Raw data deposition guidance (PRIDE/ProteomeXchange repository)
- Figure panels in publication-quality resolution (300 dpi PNG/TIFF formats)
Frequently Asked Questions About Succinylation Proteomics Analysis
Q: What is the difference between succinylation and acetylation?
Three differences matter functionally. First, charge: succinylation converts lysine from +1 to −1 (net −2 charge reversal), while acetylation converts +1 to neutral (−1 net change). Second, size: the succinyl group (100 Da) is larger than acetyl (42 Da), creating greater steric perturbation. Third, regulation: succinylation is removed by SIRT5 (~1,000× higher activity on succinyl vs. acetyl substrates), while acetylation is removed by HDACs and SIRT1-3 — these are different enzyme families with different tissue distributions and substrate preferences.
Q: How many succinylation sites can you identify from my sample?
From mammalian cell lines, expect 3,000–5,000 sites on 1,500–2,500 proteins. From tissue samples, 2,000–4,000 sites on 1,200–2,000 proteins. Mitochondrial-enriched fractions yield 800–1,500 sites. The actual number depends on sample quality, protein input amount, and fractionation depth (more fractions = more identifications).
Q: Can you distinguish enzyme-catalyzed from non-enzymatic succinylation?
Directly, no — the chemical product is identical. However, our SIRT5-regulon classification provides an operational distinction: sites whose succinylation level changes significantly upon SIRT5 genetic or pharmacological perturbation are classified as enzyme-regulated. Sites insensitive to SIRT5 at high succinyl-CoA concentrations are candidate non-enzymatic modifications. This stratification is included in standard bioinformatics reports for projects with SIRT5 comparison conditions.
Q: What enrichment method do you use for succinylated peptides?
Immunoaffinity enrichment with anti-succinyllysine motif antibodies (PTMScan® grade). Unlike phosphorylation, for which multiple chemical enrichment methods exist (TiO2, IMAC), there are currently no efficient chemical enrichment methods for succinylation. Antibody-based enrichment is the field-standard approach and achieves enrichment specificities of 50–70% (i.e., 50–70% of enriched peptides contain at least one succinyl-K).
Q: Can you combine succinylation with other PTM analyses on the same samples?
Yes. Sequential enrichment from the same tryptic digest enables co-profiling of succinylation + acetylation (+ optionally malonylation). Each PTM type requires a separate enrichment step and LC-MS/MS run but originates from the same sample, eliminating inter-sample variability. Multi-acyl crosstalk analysis is a standard option (see Section 5).
Q: What is the typical turnaround time?
Standard projects: 4–6 weeks from sample receipt to final report. Expedited service: 3–4 weeks (additional fee applies). Large cohort studies (>20 samples) and projects requiring multi-acyl co-profiling: 6–8 weeks.
Related Services
References
- Zhang T, Wang Y, Sun Y, et al. Proteome, Lysine Acetylome, and Succinylome Identify Posttranslational Modification of STAT1 as a Novel Drug Target in Silicosis. Molecular & Cellular Proteomics. 2024;23(6):100770. doi:10.1016/j.mcpro.2024.100770
- Rardin MJ, He W, Nishida Y, et al. SIRT5 Regulates the Mitochondrial Lysine Succinylome and Metabolic Networks. Cell Metabolism. 2013;18(6):920-933. doi:10.1016/j.cmet.2013.11.013
- Yang Y, Tapias V, Acosta D, et al. Altered succinylation of mitochondrial proteins, APP and tau in Alzheimer's disease. Nature Communications. 2022;13:159. doi:10.1038/s41467-021-27572-2
- Wang H, Lu J, Chen X, Schlaepfer IR. Protein succinylation: regulating metabolism and beyond. Frontiers in Nutrition. 2024;11:1336057. doi:10.3389/fnut.2024.1336057
.jpg)

