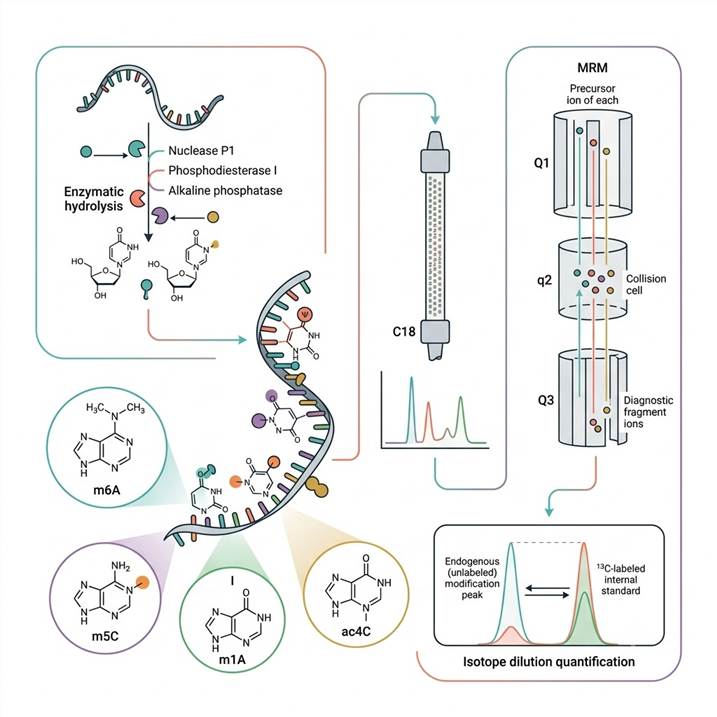
Mass Spectrometry-Based RNA Modification Quantification for Rigorous Epitranscriptomics and RNA Biology Research
Over 170 distinct chemical modifications have been identified across all RNA species — including messenger RNA (mRNA), transfer RNA (tRNA), ribosomal RNA (rRNA), small nuclear RNA (snRNA), and non-coding RNA — collectively constituting the epitranscriptome. These modifications regulate RNA stability, translation efficiency, splicing, localization, and immune recognition, and their dysregulation is increasingly implicated in cancer, neurological disorders, metabolic disease, and viral infections. Accurate quantification of RNA modification abundance is a prerequisite for understanding their biological functions, establishing their roles in disease pathogenesis, and developing RNA modification-based biomarkers and therapeutic strategies. However, RNA modification quantification presents unique analytical challenges: modifications occur at substoichiometric levels (typically 0.1–10% of total nucleosides), many modifications are chemically labile or isomeric, and the dynamic range between the most abundant (pseudouridine, m⁷G) and least abundant (m¹A, ac⁴C) modifications spans several orders of magnitude.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) addresses these challenges through a combination of high-resolution chromatographic separation, sensitive and specific mass spectrometric detection, and isotope dilution-based absolute quantification. In this workflow, RNA is first enzymatically hydrolyzed to individual nucleosides (using nuclease P1, phosphodiesterase, and alkaline phosphatase), separated by reverse-phase HPLC (typically C18 with volatile ion-pairing or aqueous-organic gradients), and detected by tandem mass spectrometry operating in either targeted (MRM/PRM) or untargeted (full-scan) acquisition modes. Targeted MRM assays provide the highest sensitivity and specificity for known modifications of interest, while untargeted full-scan analysis enables discovery of unexpected or novel modifications. The inclusion of isotopically labeled internal standards (¹³C- or ²H-labeled nucleosides) at known concentrations enables absolute molar quantification (expressed as fmol of modification per μg of RNA), permitting direct comparison across samples, experiments, and laboratories — a critical advantage over relative quantification methods. For comprehensive RNA modification analysis at the sequencing level, our mRNA Modification LC-MS Analysis and tRNA Modification LC-MS Analysis services provide RNA-species-specific modification profiling with optimized hydrolysis and enrichment protocols.
Find Your Solution: Research Goal → RNA Modification Quantification Strategy
| Your Research Goal |
Recommended Approach |
Key Techniques |
| Targeted absolute quantification of specific RNA modifications (m⁶A, Ψ, m⁵C, m¹A, m⁷G) in mRNA or total RNA across experimental conditions |
Isotope dilution LC-MRM/MS with ¹³C-labeled internal standards |
RNA hydrolysis to nucleosides, C18 reverse-phase LC separation, triple quadrupole MS in MRM mode, isotope ratio-based absolute quantification, normalization to unlabeled internal standard response |
| Comprehensive untargeted profiling of 40+ modified ribonucleosides in total RNA for epitranscriptome-wide discovery |
High-resolution LC-QTOF or LC-Orbitrap full-scan analysis with data-dependent acquisition |
Full RNA hydrolysis, HILIC or C18 LC separation, high-resolution accurate mass MS with MS/MS fragmentation, nucleoside database matching, relative abundance comparison across conditions |
| RNA modification biomarker discovery in clinical tissue or biofluid samples (plasma, serum, urine) |
High-sensitivity LC-MRM/MS with wide dynamic range and matrix-optimized sample preparation |
Protein precipitation or solid-phase extraction, C18 LC-MRM/MS with optimized ion transitions, matrix-matched calibration curves, internal standard normalization, multi-analyte panel quantification |
| Quality control of modified nucleoside content in mRNA therapeutics (IVT mRNA, circRNA, saRNA) |
Targeted LC-MRM/MS with GMP-compatible documentation |
RNase-based oligonucleotide digestion or complete nucleoside hydrolysis, LC-MRM/MS for incorporated modification percentage, purity assessment, batch-to-batch comparability, ICH M10-aligned method validation |
| Quantification of low-abundance RNA modifications (m¹A, ac⁴C, m³C, hm⁵C) requiring enhanced sensitivity |
Immunoaffinity enrichment or chemical labeling coupled with nanoLC-MRM/MS |
Anti-modification antibody enrichment, chemical derivatization for ionization enhancement, nanoLC-MRM/MS with microflow chromatography, signal amplification through selective modification tagging |
| Multi-omics integration: correlating RNA modification abundance with transcript expression or proteomics data |
Parallel RNA modification LC-MS and RNA-seq sample processing from the same biological specimen |
Splitting RNA extract for parallel LC-MS modification quantification and RNA-seq library preparation, normalization to spike-in controls, multi-omic correlation analysis, Systems biology data integration |
Complementary LC-MS/MS Platforms for Targeted and Untargeted RNA Modification Quantification
Our RNA modification quantification service portfolio encompasses three complementary LC-MS/MS platforms, each optimized for specific sensitivity, coverage, throughput, and quantitative accuracy requirements. Platform selection depends on the research question, RNA sample availability, modification targets of interest, and desired quantification depth.
Triple Quadrupole (QqQ) LC-MRM/MS for Targeted Absolute Quantification with Gold-Standard Sensitivity and Specificity
For targeted quantification of known RNA modifications with the highest sensitivity and specificity, we employ triple quadrupole mass spectrometers operating in multiple reaction monitoring (MRM) mode. In this configuration, the first quadrupole (Q1) selects the precursor ion corresponding to the nucleoside of interest (e.g., m⁶A at m/z 282.1 → 150.1), the second quadrupole (q2) serves as a collision cell for fragmentation, and the third quadrupole (Q3) selects a specific fragment ion, providing two stages of mass filtering that effectively eliminates chemical noise from complex biological matrices. Isotope dilution quantification using ¹³C- or ²H-labeled internal standards (e.g., [¹³C₅]-m⁶A, [¹³C₅]-pseudouridine, [D₃]-m⁵C) achieves absolute quantification with limits of detection as low as 0.1–1 fmol on-column and dynamic range spanning 3–5 orders of magnitude. Parallel reaction monitoring (PRM) on quadrupole-Orbitrap instruments provides an alternative targeted approach with high-resolution full MS/MS scans for enhanced specificity in complex sample matrices. For broader RNA modification analysis beyond individual targets, our DNA/RNA Modification LC-MS Analysis service provides comprehensive modification detection across both DNA and RNA nucleosides in a single analytical run.
High-Resolution Quadrupole Time-of-Flight (QTOF) and Orbitrap LC-MS for Untargeted RNA Modification Discovery
For comprehensive untargeted profiling of the RNA modification landscape, we employ high-resolution accurate mass (HRAM) mass spectrometry using QTOF and Orbitrap platforms. Full-scan acquisition at resolving power of 60,000–120,000 (FWHM at m/z 200) enables detection of all ionizable nucleosides without pre-selection of target analytes, providing a global view of the RNA modification landscape. High-resolution mass measurements (<3 ppm mass accuracy) enable unambiguous assignment of elemental compositions to unknown nucleosides, and data-dependent or data-independent MS/MS acquisition provides structural information for modification identification. This untargeted approach is particularly valuable for discovery-phase research aimed at identifying novel RNA modifications, characterizing modification changes under non-canonical biological conditions, or comprehensively profiling modifications in understudied RNA species. Identified modification features can be subsequently validated and absolutely quantified using targeted MRM assays in follow-up studies. For oligonucleotide-level RNA modification detection, our Oxidative DNA/RNA Damage Assay provides complementary LC-MS/MS analysis of oxidatively damaged nucleosides at both nucleoside and oligonucleotide levels.
Nanoflow and Microflow LC-MS/MS for High-Sensitivity Quantification from Limited RNA Input
For research projects with limited RNA material — including clinical biopsy samples, laser capture microdissected tissue, rare cell populations, or single-cell-equivalent RNA amounts — our nanoflow and microflow LC-MS/MS platforms provide enhanced sensitivity through reduced chromatographic dilution and improved ionization efficiency at low flow rates (50–500 nL/min for nanoLC, 2–10 μL/min for microLC). These platforms incorporate the same MRM and high-resolution acquisition capabilities as analytical-flow systems but achieve 10–100-fold improvements in limits of detection by concentrating the analyte into a smaller elution volume and delivering it to the mass spectrometer ion source at lower flow rates that favor more efficient droplet formation and ionization. Sample requirements for nanoLC-MRM/MS quantification of abundant RNA modifications (Ψ, m⁷G, m⁶A) can be as low as 1–10 ng of total RNA, enabling modification analysis from samples where RNA quantity is the primary limiting factor. For dedicated analysis of specific RNA modification types, our m⁶A Modification LC-MS Analysis and DNA/RNA Modification Immunoassays services provide complementary targeted detection and orthogonal validation platforms.
Why Choose Our RNA Modification Quantification by LC-MS/MS Service
Gold-Standard Absolute Quantification with Isotope Dilution Mass Spectrometry
Our LC-MS/MS platform delivers true absolute quantification of RNA modifications using isotope dilution mass spectrometry with ¹³C- and ²H-labeled internal standards — the gold-standard approach for quantitative mass spectrometry. Unlike relative quantification methods that report fold-changes without absolute abundance, or sequencing-based methods that infer modification presence through indirect detection, our isotope dilution approach provides unambiguous molar quantification (fmol modification per μg RNA) that is directly comparable across samples, experiments, and laboratories. This absolute quantitative foundation is essential for establishing baseline modification levels, comparing results across independent studies, and supporting regulatory-relevant quality control applications.
Comprehensive RNA Modification Coverage Across All Major Modified Ribonucleoside Classes
Our optimized LC-MS/MS platform simultaneously detects and quantifies 40+ modified ribonucleosides spanning all major RNA modification classes — including base methylations (m⁶A, m⁵C, m¹A, m⁷G, m³C, m²G, m¹G, m⁵U, m³U), pseudouridine (Ψ) and its 2'-O-methylated derivative (Ψm), acetylated modifications (ac⁴C), deaminated modifications (inosine, I), 2'-O-methylated nucleosides (Am, Gm, Cm, Um), and hypermodified tRNA nucleosides (t⁶A, ms²i⁶A, i⁶A, Q, oQ, wybutosine, mcm⁵U, mcm⁵s²U, ncm⁵U, ncm⁵Um) — in a single analytical injection, providing the broadest practical coverage for comprehensive RNA modification quantification.
Validated Multi-RNA-Species Workflows with Optimized Sample Preparation Protocols
We have developed and validated dedicated sample preparation and LC-MS/MS workflows for each major RNA species type — mRNA (purified by polyA selection or rRNA depletion), total RNA, tRNA (isolated by size exclusion or guanidinium-based methods), rRNA, small RNA (miRNA, siRNA), and therapeutic mRNA (IVT, circRNA, saRNA) — with RNA hydrolysis protocols optimized for each RNA type to ensure complete digestion without modification degradation. Sample requirements are as low as 10–100 ng for targeted MRM quantification of abundant modifications in total RNA, and 100 ng–1 μg for comprehensive profiling of 40+ modifications, with published validation data demonstrating inter-day precision of <15% CV and accuracy of 85–115% for most target modifications.
Epitranscriptomics-Integrated Bioinformatics with Multi-Omic Data Correlation
Our RNA modification quantification service is complemented by a dedicated bioinformatics pipeline that not only processes raw LC-MS data for peak integration, isotope ratio calculation, and modification identification, but also provides epitranscriptomics-focused data analysis including modification abundance normalization, differential modification analysis across experimental groups, correlation analysis with matched transcriptomics or proteomics data, and pathway-level interpretation of modification changes. This multi-omic integration capability distinguishes our service from standard LC-MS providers, enabling researchers to connect RNA modification changes to transcriptional and proteotypic functional outcomes.
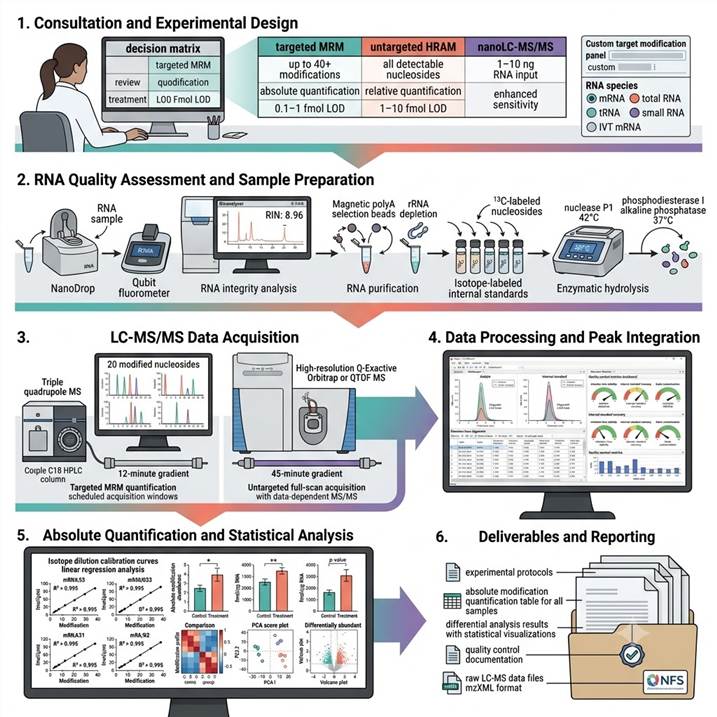
Workflow: From RNA Sample to Absolutely Quantified Modification Data
Step 1: Consultation and Experimental Design
We consult with you to define the RNA modification quantification strategy based on RNA species of interest (mRNA, total RNA, tRNA, small RNA, therapeutic RNA), target modifications (targeted MRM panel or untargeted full-scan profiling), sample type (cell lysate, tissue RNA, biofluid, purified RNA), quantification approach (absolute with isotope standards or relative comparison), experimental design including biological replicates (minimum 3 recommended), comparison groups, and statistical power requirements. We provide a detailed project proposal with platform recommendations and expected modification coverage.
Step 2: RNA Quality Assessment and Sample Preparation
RNA samples are assessed for quantity (NanoDrop, Qubit), purity (A260/A280, A260/A230 ratios), and integrity (RIN value by Bioanalyzer or TapeStation). Depending on the RNA species of interest, samples undergo purification steps: polyA selection or rRNA depletion for mRNA analysis, size exclusion for tRNA isolation, or total RNA direct analysis. Isotope-labeled internal standards (¹³C- or ²H-labeled nucleosides) are added at known concentrations to each sample before hydrolysis to enable absolute quantification. RNA is enzymatically hydrolyzed using nuclease P1, phosphodiesterase I, and alkaline phosphatase to release individual nucleosides.
Step 3: LC-MS/MS Data Acquisition
Hydrolyzed nucleoside samples are analyzed by our optimized LC-MS/MS platform. For targeted quantification: samples are separated by C18 reverse-phase HPLC with aqueous-organic gradient and analyzed by triple quadrupole MS in MRM mode with scheduled acquisition windows for each modification-internal standard pair. For untargeted profiling: samples are analyzed by high-resolution QTOF or Orbitrap MS in full-scan mode with data-dependent MS/MS acquisition. Each analytical batch includes calibration standards, quality control samples, and blank injections for system suitability monitoring.
Step 4: Data Processing and Peak Integration
Raw LC-MS data are processed using dedicated software pipelines. For targeted MRM data: chromatographic peaks are integrated, retention times aligned, and peak area ratios (analyte/internal standard) calculated for each modification. For untargeted HRAM data: features are detected, aligned across samples, and identified by accurate mass matching (<3 ppm) and MS/MS spectral matching against our in-house nucleoside database and public repositories (METLIN, mzCloud, GNPS). Quality control metrics including retention time stability, internal standard response, and blank contamination are evaluated for each analytical batch.
Step 5: Absolute Quantification and Statistical Analysis
Absolute modification abundance (fmol modification per μg RNA) is calculated using isotope dilution calibration curves constructed from authentic nucleoside standards and their corresponding isotope-labeled internal standards. Differential modification analysis between experimental groups is performed using appropriate statistical tests (Student's t-test, ANOVA, or non-parametric alternatives) with multiple testing correction (Benjamini-Hochberg FDR). Principal component analysis and hierarchical clustering are applied for global modification pattern visualization. Correlation analysis with matched transcriptomics or proteomics data is performed for multi-omic integration studies.
Step 6: Deliverables and Reporting
Comprehensive RNA modification quantification report including: complete experimental protocols with RNA preparation, hydrolysis, and LC-MS/MS methods, absolute modification abundance values (fmol/μg RNA) for all quantified modifications with retention times and MRM transitions, differential modification analysis results with statistical testing and visualization (volcano plots, heat maps, box plots), quality control metrics with internal standard recovery and system suitability data, and multi-omic correlation analysis results for integrated studies. All raw data (LC-MS files, peak integration results) are provided in standard formats for regulatory reference and independent re-analysis.
Applications in RNA Modification Quantification Research
LC-MS/MS-based RNA modification quantification has become the reference method for accurate measurement of modified ribonucleoside abundance across diverse research applications. The following application areas represent the most active and impactful use cases for our quantitative RNA modification analysis platform.
Epitranscriptomics Discovery: Comprehensive Modification Landscape Mapping Across Biological Conditions
LC-MS/MS-based untargeted profiling of 40+ modified ribonucleosides provides a comprehensive quantitative view of the epitranscriptome across different biological states, developmental stages, or disease conditions. By comparing total RNA or mRNA modification profiles between control and experimental groups, researchers can identify modification-level changes that correlate with phenotypic differences, discover co-regulated modification patterns, and prioritize specific modifications for mechanistic follow-up studies. The ability to simultaneously quantify modifications from multiple classes (methylations, pseudouridylation, acetylation, deamination) in a single analysis provides an integrated view of epitranscriptomic regulation that is not achievable by modification-specific sequencing methods. For targeted analysis of specific modification types, our mRNA Modification LC-MS Analysis service provides dedicated mRNA modification profiling with polyA-selected RNA.
Disease Biomarker Discovery: Quantifying RNA Modification Signatures in Clinical Biospecimens
RNA modifications — particularly modified ribonucleosides that are released into circulation during RNA turnover — have emerged as promising biomarkers for cancer, neurological disorders, cardiovascular disease, and metabolic conditions. LC-MS/MS quantification of modified ribonucleosides in plasma, serum, urine, and tissue biopsies enables the identification and validation of RNA modification-based biomarker panels with diagnostic, prognostic, or predictive value. The absolute quantification capability of isotope dilution LC-MS/MS is particularly important for biomarker applications, as it enables establishment of reference ranges, cross-study comparability, and longitudinal monitoring with quantitative confidence not achievable by relative quantification approaches.
mRNA Therapeutic Quality Control: Quantifying Modified Nucleoside Content and Purity
The incorporation of modified nucleosides — particularly N¹-methylpseudouridine (m¹Ψ) and pseudouridine (Ψ) — into in vitro transcribed (IVT) mRNA therapeutics is critical for reducing immunogenicity, enhancing translation efficiency, and improving mRNA stability. LC-MS/MS-based quantification provides the definitive analytical method for measuring the percentage replacement of natural uridine with modified uridine analogs in therapeutic mRNA, assessing batch-to-batch consistency, detecting unintended modifications introduced during IVT, and characterizing base composition. Our LC-MS/MS platform supports regulatory-relevant quality control under ICH M10-aligned method validation guidelines, providing the quantitative rigor required for mRNA therapeutic development and manufacturing. For broader RNA damage assessment, our Oxidative DNA/RNA Damage Assay provides complementary LC-MS/MS analysis of oxidatively damaged nucleoside species.
RNA Modification Enzyme and Inhibitor Studies: Quantifying Substrate-Product Relationships
Studying RNA methyltransferases (METTL3/METTL14, NSUN2, TRMTs), demethylases (FTO, ALKBH5), pseudouridine synthases (PUS1-7, DKC1), and their inhibitors requires accurate quantification of substrate and product modification levels to determine enzyme activity, inhibition potency, and target engagement. LC-MS/MS provides direct quantification of both the unmodified and modified nucleoside species, enabling precise determination of modification stoichiometry, enzyme kinetic parameters (Km, kcat), and inhibitor IC₅₀ values — measurements that are essential for both basic mechanistic studies and drug development programs targeting RNA modification enzymes. The ability to quantify multiple modifications simultaneously also enables assessment of inhibitor selectivity across the modification landscape.
tRNA Modification Biology: Quantifying the Complex tRNA Modification Landscape and Its Dynamics
Transfer RNA (tRNA) contains the highest density and diversity of RNA modifications — with an average of 13 modifications per tRNA molecule, including complex hypermodified nucleosides (t⁶A, ms²i⁶A, Q, wybutosine, mcm⁵U, mcm⁵s²U) that are unique to tRNA and are essential for decoding accuracy, reading frame maintenance, and tRNA stability. LC-MS/MS quantification of tRNA modifications provides a comprehensive quantitative view of the tRNA modification landscape, enabling studies of tRNA modification dynamics in response to cellular stress, nutrient availability, differentiation signals, and disease states. Our optimized tRNA hydrolysis and LC-MS/MS workflows are specifically designed to preserve and detect the full diversity of tRNA modifications, including acid-labile and hypermodified species that are challenging for standard RNA hydrolysis protocols. For dedicated tRNA-focused analysis, our tRNA Modification LC-MS Analysis service provides optimized tRNA purification and modification profiling workflows.
Case Study: LC-MS/MS-Based Absolute Quantification of Methylated Guanosine and Uridine Modifications in S. cerevisiae mRNA Reveals Translation Elongation Regulation
In a 2023 study published in RSC Chemical Biology (CC BY 3.0), Jones et al. developed and applied an optimized LC-MS/MS pipeline for absolute quantification of methylated ribonucleoside modifications in purified yeast mRNA, establishing a rigorous quantitative framework for studying the functional roles of mRNA modifications in translation regulation.
Background: While over 170 RNA modifications had been catalogued across all RNA species, the presence and functional significance of internal mRNA modifications beyond m⁶A — particularly methylated guanosine and uridine residues — were poorly understood. The limited detection of these modifications in previous studies was potentially attributable to insufficient mRNA purification stringency (contamination by highly modified rRNA and tRNA) rather than true absence from mRNA. The authors hypothesized that improved mRNA purification combined with sensitive LC-MS/MS quantification would reveal a broader landscape of mRNA modifications and enable functional studies of their roles in translation.
Approach: The authors implemented a multi-step mRNA purification strategy combining polyA selection with RNase H-based rRNA depletion to achieve >99% mRNA purity (verified by Bioanalyzer and RT-qPCR), eliminating modification carryover from highly modified rRNA and tRNA. Purified mRNA was enzymatically hydrolyzed to individual nucleosides (nuclease P1, phosphodiesterase I, alkaline phosphatase) and analyzed by LC-MS/MS using both untargeted high-resolution full-scan acquisition (Q-Exactive Orbitrap, C18 column, 45-minute gradient) for modification discovery and targeted PRM acquisition with isotope-labeled internal standards for absolute quantification. The LC-MS/MS method was validated for 50 ribonucleosides — including 13 methylated guanosine and uridine modifications and their corresponding natural unmodified nucleosides — achieving the lowest limits of detection reported for ribonucleoside modification analysis at that time.
Key Findings:
- Identification of four new mRNA modifications: The improved purification and LC-MS/MS pipeline enabled detection and absolute quantification of 13 methylated ribonucleoside modifications in S. cerevisiae mRNA, including four modifications not previously identified in mRNA: 1-methylguanosine (m¹G), N²-methylguanosine (m²G), N²,N²-dimethylguanosine (m²₂G), and 5-methyluridine (m⁵U), expanding the known mRNA modification landscape beyond the established m⁶A, m⁵C, and Ψ
- Enzyme assignment and genetic validation: Using a systematic knockout library of known and predicted RNA methyltransferase genes, the authors identified the responsible methyltransferase enzymes for each modification: Trm10 for m¹G in mRNA, Trm11 for m²G, Trm1 for m²₂G, and Trm2 for m⁵U, demonstrating that these modifications are introduced by the same enzymes that deposit them in tRNA, revealing an unanticipated cross-RNA-species modification pathway
- Absolute quantification across conditions: Isotope dilution LC-MS/MS with ¹³C-labeled internal standards provided absolute quantification of each modification (fmol/μg mRNA), establishing baseline abundance levels ranging from 0.01–0.5% modification fraction relative to unmodified nucleosides, and demonstrating that modification levels are dynamically regulated in response to growth phase and nutrient availability
- Functional impact on translation elongation: In vitro translation assays using reporter mRNAs with site-specifically incorporated m¹G, m²G, and m⁵U demonstrated that these modifications impede translation elongation in a position-dependent manner — with modifications in the codon body causing greater elongation inhibition than those in the 5′ UTR — providing the first direct evidence that these newly identified mRNA modifications have functional consequences for protein synthesis
- Methodological validation: The LC-MS/MS pipeline was rigorously validated with limits of detection of 0.5–5 fmol on-column for modified ribonucleosides, inter-day precision of <15% CV, and linear dynamic range spanning 3–4 orders of magnitude, establishing a reference analytical framework for absolute quantification of mRNA modifications by LC-MS/MS that has been widely adopted by the epitranscriptomics field
Significance: This study established that the yeast mRNA modification landscape is substantially more complex than previously recognized, identified four new mRNA modifications with their responsible methyltransferase enzymes, and provided the first direct evidence that methylated guanosine and uridine modifications in mRNA codons impede translation elongation. The optimized LC-MS/MS quantification pipeline — combining stringent mRNA purification, comprehensive nucleoside coverage, and isotope dilution absolute quantification — has become a reference methodological framework for LC-MS/MS-based RNA modification quantification studies. The approach is directly transferable to our service platform, where we similarly employ optimized RNA purification, comprehensive modification panels, and isotope dilution absolute quantification to deliver rigorous quantitative RNA modification data.
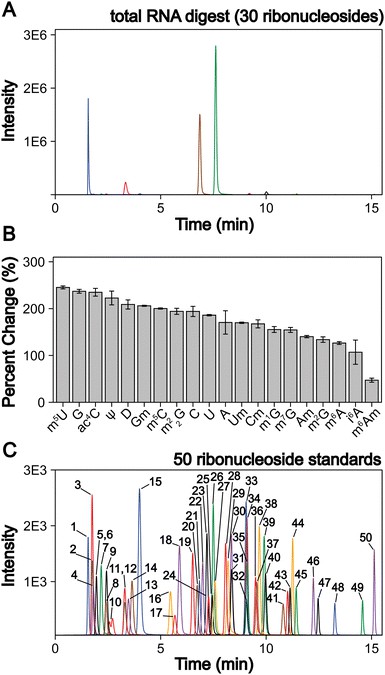
Adapted from Jones et al. (2023). Methylated guanosine and uridine modifications in S. cerevisiae mRNAs modulate translation elongation. RSC Chemical Biology 4:363–378. (CC BY 3.0)
Representative Results: RNA Modification Quantification Data Outputs and Platform Performance
Our RNA modification quantification platform delivers integrated data packages combining absolute quantification values, quality-controlled raw data, and comprehensive statistical analysis for publication-ready reporting. The representative data below illustrates the typical output quality and platform specifications achieved across our targeted and untargeted LC-MS/MS workflows.
Platform Performance Specifications
| Performance Parameter |
Targeted MRM/PRM LC-MS/MS |
Untargeted HRAM LC-MS/MS |
| Modification coverage |
Up to 40+ targeted modifications per injection (custom panel design) |
All detectable nucleosides (untargeted, 40–60+ per sample) |
| Quantification type |
Absolute (isotope dilution with ¹³C/²H internal standards) |
Relative (label-free peak area comparison) |
| Limit of detection |
0.1–1 fmol on-column (modification-dependent) |
1–10 fmol on-column (modification-dependent) |
| Dynamic range |
3–5 orders of magnitude |
2–4 orders of magnitude |
| Inter-day precision |
<15% CV for most modifications |
<25% CV for most modifications |
| Sample requirement (total RNA) |
10–100 ng (abundant modifications); 100 ng–1 μg (comprehensive panel) |
100 ng–1 μg total RNA |
| RNA species compatibility |
mRNA, total RNA, tRNA, rRNA, small RNA, IVT mRNA |
Total RNA, mRNA, tRNA |
| Throughput (samples/week) |
40–80 samples (12-minute gradient); 20–40 (30-minute gradient) |
15–25 samples per instrument |
| Reporting format |
fmol/μg RNA, modification fraction (%), fold-change, statistics |
Peak area, normalized abundance, fold-change, PCA, clustering |
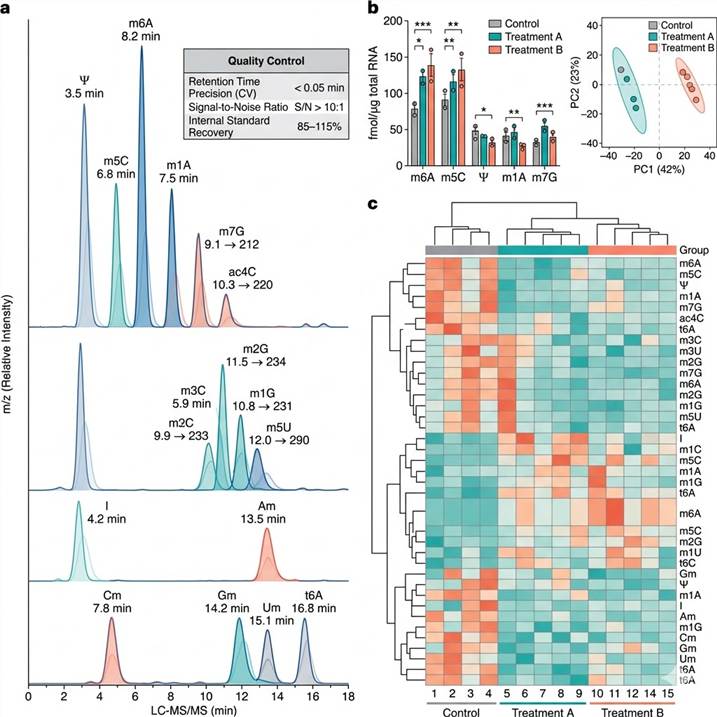
Representative data outputs from our RNA Modification Quantification by LC-MS/MS platform. Left: Multiplexed MRM chromatogram of 20 modified ribonucleosides with internal standards. Center: Absolute quantification comparison across experimental groups. Right: Global modification profile heat map and PCA clustering.
Key data deliverables included in every RNA modification quantification project:
- Absolute modification quantification data package — Complete table of all quantified modifications with absolute abundance values (fmol/μg RNA), retention times, MRM transitions or accurate mass measurements, limit of detection, and internal standard recovery for each analyzed sample, with chromatographic peak quality flags for data quality assessment
- Comparative and statistical analysis package — Differential modification analysis results with appropriate statistical testing (Student's t-test, ANOVA, Mann-Whitney U, or mixed-effects models) and multiple testing correction (Benjamini-Hochberg FDR), with volcano plots, bar charts, box plots, and heat maps for data visualization, plus PCA and hierarchical clustering for global modification pattern analysis
- Quality control documentation — Full quality control report including system suitability test results (retention time stability, mass accuracy, signal intensity), calibration curve performance (linearity, accuracy, precision), internal standard recovery across all samples, blank contamination assessment, and inter-batch normalization metrics
- Multi-omic integration data — Correlation analysis between RNA modification abundance and matched transcript expression, protein expression, or epitranscriptomic sequencing data (when provided), with visualization of significant correlations and pathway-level interpretation of coordinated modification-expression changes
- Methods documentation — Complete protocols for RNA purification, hydrolysis, LC-MS/MS acquisition, and data analysis, formatted for publication methods sections and regulatory reference (ICH M10-aligned where applicable)
Related Services
Our RNA modification quantification by LC-MS/MS platform is part of a comprehensive DNA/RNA modification analysis service portfolio spanning LC-MS quantification, modification-specific LC-MS analysis, immunoassay-based detection, adductomics, and multi-omic integration for molecular biology and translational research programs.
- DNA/RNA Modification LC-MS Analysis — Integrated LC-MS analysis platform covering both DNA and RNA modifications in a single analytical workflow for comprehensive nucleoside modification profiling
- m⁶A Modification LC-MS Analysis — Dedicated LC-MS analysis of N⁶-methyladenosine (m⁶A) and related adenine modifications with optimized enrichment and quantification protocols
- mRNA Modification LC-MS Analysis — Targeted LC-MS/MS profiling of modified ribonucleosides in purified mRNA with polyA selection and rRNA depletion workflows
- tRNA Modification LC-MS Analysis — Specialized tRNA purification and LC-MS/MS profiling optimized for detection of hypermodified and acid-labile tRNA-specific nucleosides
- DNA/RNA Modification Immunoassays — Antibody-based colorimetric and fluorescent immunoassay platforms for targeted detection of specific DNA and RNA modifications
- Oxidative DNA/RNA Damage Assay — Targeted LC-MS/MS quantification of oxidatively damaged nucleoside species in DNA and RNA for oxidative stress research and toxicology studies
FAQs
How does LC-MS/MS-based RNA modification quantification differ from RNA sequencing-based methods?
LC-MS/MS directly detects and quantifies modified ribonucleosides at the molecular level based on their chemical properties (mass, retention time, fragmentation pattern), providing unambiguous identification and absolute molar quantification. RNA sequencing-based methods infer modification presence indirectly through reverse transcription signatures, antibody enrichment biases, or bioinformatic prediction, each of which introduces specific limitations: many modifications (pseudouridine, m¹A) cause only partial reverse transcription stops, antibody enrichment is subject to sequence context bias, and bioinformatic predictions require independent validation. LC-MS/MS is the gold standard for accurate modification quantification, while sequencing methods provide transcriptome-wide modification site location information that LC-MS/MS cannot provide. The two approaches are complementary — LC-MS/MS provides quantitative accuracy and breadth, while sequencing provides site-level resolution.
What types of RNA modifications can be quantified by LC-MS/MS?
Our platform can detect and quantify 40+ modified ribonucleosides spanning all major RNA modification classes, including: base methylations (m⁶A, m⁵C, m¹A, m⁷G, m³C, m²G, m¹G, m⁵U, m³U), pseudouridine (Ψ) and its 2'-O-methylated derivative (Ψm), acetylated modifications (ac⁴C), deaminated modifications (inosine, I), 2'-O-methylated nucleosides (Am, Gm, Cm, Um), dihydrouridine (D), and hypermodified tRNA nucleosides (t⁶A, ms²i⁶A, i⁶A, Q, oQ, wybutosine, mcm⁵U, mcm⁵s²U, ncm⁵U, ncm⁵Um). Custom panels can be designed for client-specific modifications or for quantification of modified nucleotides incorporated into therapeutic RNAs (e.g., N¹-methylpseudouridine in IVT mRNA).
What is the minimum RNA amount required for LC-MS/MS quantification?
Sample requirements depend on the RNA species, target modifications, and quantification approach. For targeted MRM quantification of abundant modifications (Ψ, m⁷G, m⁶A) in total RNA, as little as 10–100 ng is sufficient. For comprehensive targeted quantification of 40+ modifications, 100 ng–1 μg of total RNA is recommended. For untargeted high-resolution full-scan profiling, 100 ng–1 μg of total RNA is typically required. For mRNA-specific analysis, 100–500 ng of purified mRNA (corresponding to 5–25 μg of total RNA starting material) is recommended. For tRNA-specific analysis, 50–200 ng of purified tRNA is sufficient. Our nanoflow LC-MS/MS platform can extend detection to as low as 1–10 ng of total RNA for abundant modifications in limited-sample scenarios.
How is absolute quantification of RNA modifications achieved?
Absolute quantification is achieved through isotope dilution mass spectrometry. Known quantities of isotope-labeled internal standards (¹³C- or ²H-labeled modified nucleosides) are added to each RNA sample prior to enzymatic hydrolysis. The ratio of the unlabeled (endogenous) modification peak area to the labeled (internal standard) peak area is measured by LC-MS/MS, and the absolute amount of each modification is calculated by reference to a calibration curve constructed from authentic nucleoside standards analyzed under identical conditions. Results are reported as fmol of modification per μg of RNA, or alternatively as modification fraction (percentage of modified nucleoside relative to total unmodified nucleoside). This approach compensates for matrix effects, extraction losses, and ionization efficiency variations, providing accurate and reproducible absolute quantification that is directly comparable across samples, experiments, and laboratories.
Can LC-MS/MS distinguish between isomeric RNA modifications?
Yes — LC-MS/MS can distinguish between many isomeric RNA modifications through a combination of chromatographic separation and differential fragmentation. For example, m⁶A and m¹A (both C₁₁H₁₅N₅O₅, monoisotopic mass 297.1073 Da) are resolved by C18 reverse-phase chromatography under optimized gradient conditions, and their fragmentation spectra show distinct patterns (m⁶A produces the characteristic base ion at m/z 150.077 while m¹A generates diagnostic ions at m/z 136.062 and 109.051 due to Dimroth rearrangement). Similarly, m⁵C and m³C are distinguished by retention time and differential fragmentation. However, some isomeric modifications (e.g., Cm and m³C, which share the same accurate mass and similar retention behavior) may require additional method optimization, orthogonal separation (HILIC), or MS³ fragmentation for unambiguous assignment. We validate modification identification using authentic standards wherever possible and report identification confidence levels for each modified nucleoside.
How do you prevent RNA degradation and modification loss during sample processing?
Preservation of RNA integrity and modification stability is critical for accurate quantification. Our protocols incorporate several measures: (1) all RNA samples are processed under RNase-free conditions with dedicated equipment and reagents; (2) RNA integrity is verified by Bioanalyzer or TapeStation before and after purification; (3) enzymatic hydrolysis conditions (pH, temperature, incubation time) are optimized to ensure complete digestion while minimizing modification degradation — particularly for acid-labile modifications (m¹A, m³C) and alkali-sensitive modifications (ac⁴C); (4) isotope-labeled internal standards added at the earliest possible step compensate for any post-lysis modification changes; (5) samples are analyzed promptly after hydrolysis or stored at −80°C for short-term storage; (6) degradation markers (increase in unmodified nucleoside peak area ratios) are monitored as quality control metrics for each sample batch.
How is RNA modification quantification data analyzed and reported?
Raw LC-MS data are processed through a dedicated bioinformatics pipeline. For targeted MRM/PRM data: raw files are imported into quantitative analysis software (Skyline, MultiQuant, TraceFinder), chromatographic peaks are automatically integrated with manual inspection for quality control, retention times are aligned across the batch, and peak area ratios (analyte/internal standard) are calculated and exported for statistical analysis. For untargeted HRAM data: features are detected and aligned using dedicated software (ProteoWizard, MZmine, Compound Discoverer), identified by accurate mass matching and MS/MS spectral library searching, and normalized for cross-sample comparison. Statistical analysis is performed in R or Python using packages for differential modification analysis, multiple testing correction, PCA, and hierarchical clustering. Data are delivered as absolute modification abundance tables (fmol/μg RNA) with quality metrics, statistical results with visualization plots, and raw LC-MS data files in standard open formats (mzXML, mzML) for independent re-analysis and regulatory submission.
References
- Jones JD, Franco MK, Smith TJ, Snyder LR, Anders AG, Ruotolo BT, Kennedy RT, Koutmou KS. Methylated guanosine and uridine modifications in S. cerevisiae mRNAs modulate translation elongation. RSC Chemical Biology. 2023;4:363–378.
- Borland K, et al. Toward standardized epitranscriptome analytics: inter-laboratory comparison of LC-MS/MS-based RNA modification quantification. Nucleic Acids Research. 2025;gkaf895.
- Mauer J, et al. Global analysis by LC-MS/MS of N⁶-methyladenosine and inosine in mRNA reveal complex incidence. RNA. 2025;31(4):514–528.
For research use only. Not for use in diagnostic procedures.