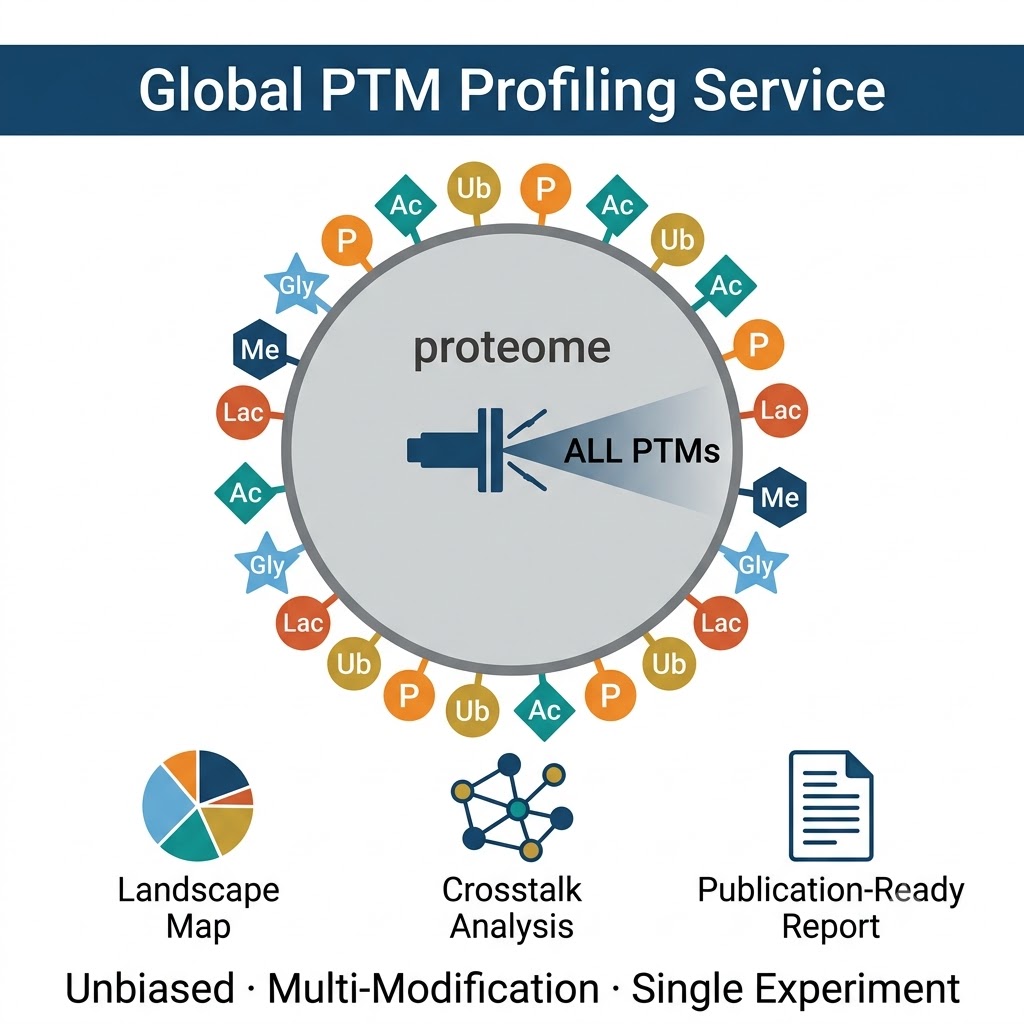
What Is Global PTM Profiling — and When Is It the Right Experiment?
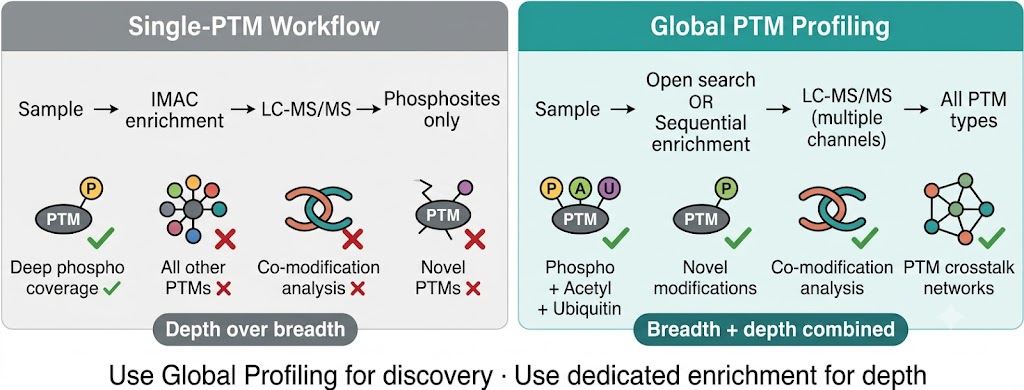
The conventional PTM proteomics paradigm applies a single modification-specific enrichment protocol — IMAC for phosphorylation, diGLY antibody IP for ubiquitylation, pan-acetyllysine antibody for acetylation — to concentrate one class of modified peptides before LC-MS/MS analysis. This approach delivers outstanding depth for the targeted modification type, but it is structurally blind to everything else. A phosphoproteomics experiment tells you about phosphorylation. It tells you nothing about whether the same proteins are simultaneously ubiquitinated, acetylated, or carrying acyl marks that may be functionally more important in your specific biological context.
Global PTM Profiling resolves this limitation through two complementary strategies — open-search unbiased modification discovery and sequential multi-PTM enrichment — that together survey the full modification space of a sample without predetermining which PTMs are interrogated. The result is a modification landscape: an integrated view of which proteins are modified, by which modification types, on which specific residues, and in which combinations — from a single project.
Use Global PTM Profiling When:
- You are studying an organism, cell type, or disease state where the PTM landscape has not been characterized and you do not know which modifications are predominant or regulated.
- You have a biological system that undergoes major perturbation (drug treatment, stress, differentiation, infection) and you want an unbiased view of the full PTM response — not just the modification type you expected to change.
- You need a publication-grade PTM dataset that demonstrates breadth of modification coverage — not just phosphoproteomics, but a multi-modification characterization of your system.
- You want to identify novel PTM types or modification combinations on specific proteins of interest, including unexpected modifications introduced by metabolic conditions, drug treatment, or cellular stress.
- You are designing a targeted follow-up experiment (DIA quantification, PRM verification) and need a comprehensive site list to define the target universe — because targeted experiments can only confirm what you already know to look for.
What Global Profiling Reveals That Single-PTM Workflows Miss:
- Co-modification patterns: proteins carrying multiple modification types simultaneously — the combinatorial PTM code on histones, transcription factors, and signaling regulators that single-PTM enrichments cannot detect because each enrichment is performed separately on separate samples.
- Unexpected modification types: novel acylation marks, non-enzymatic modifications (glycation, carbonylation, deamidation), drug-induced covalent adducts, or metabolite-derived modifications that no single-PTM workflow would identify because no enrichment resin exists for them.
- Modification crosstalk networks: by identifying multiple modification types on the same proteins in the same experiment, global profiling directly enables PTM crosstalk analysis — identifying regulatory relationships between phosphorylation and ubiquitylation, acetylation and methylation, or any other modification pair on shared substrates.
- Modification type distribution: the relative frequency of different modification classes across the proteome — revealing which modifications dominate the regulatory landscape of your biological system, and where to focus modification-specific deep-dive experiments for maximum biological yield.
Global Profiling vs. Single-PTM Deep Profiling — Choosing the Right Approach:
| Criterion |
Global PTM Profiling |
Single-PTM Deep Profiling |
| Modification coverage |
All detectable types simultaneously |
One modification type per experiment |
| Site depth per PTM type |
Moderate (unenriched) to high (sequential enrichment) |
Maximum (dedicated enrichment) |
| Novel modification discovery |
Yes — open search detects unanticipated PTMs |
No — limited to predefined variable modifications |
| Co-modification analysis |
Yes — from the same sample |
No — requires separate experiments |
| Best stage |
Early discovery, hypothesis generation, landscape mapping |
Deep quantitative profiling after target modification is known |
| Sample input |
Moderate (1–5 mg per sample) |
Higher (5–20 mg for enrichment-based) |
Global PTM Profiling Strategies
We offer three complementary global profiling strategies — each covering a different part of the modification space and suited to different experimental goals. Projects can use a single strategy or a combination, depending on the breadth and depth required.
Strategy 1 — Open-Search PTM Discovery
Unbiased, library-free identification of known and unanticipated modifications in unenriched samples using wide precursor mass tolerance or tag-based open search algorithms (FragPipe/MSFragger, MetaMorpheus G-PTM-D, PTM-Shepherd). The search tolerates a mass window of ±500–1000 Da around each peptide, identifying all mass additions consistent with known or novel modifications at each residue.
What it detects: all enzymatic PTMs present at sufficient abundance in the unenriched sample — phosphorylation, acetylation, methylation, deamidation, oxidation, carbamylation, glycation, and modification types not anticipated in a conventional closed search. Also identifies sample preparation artifacts (incomplete alkylation, oxidized methionine) that affect data quality.
Depth: reflects the modification stoichiometry distribution in the unenriched sample — high-stoichiometry modifications (abundant acetylation on histones, constitutive phosphorylation on abundant proteins) are detected confidently; low-stoichiometry modifications require enrichment for detection. Open search is best understood as a modification survey tool, not a deep site-mapping tool.
Software: MSFragger (FragPipe suite) open search; PTM-Shepherd for modification type summarization and site annotation; MetaMorpheus G-PTM-D for combined curated + novel modification discovery.
Link to dedicated service: Open-Search PTM Discovery Service →
Strategy 2 — Sequential Multi-PTM Enrichment
Two to four modification-specific enrichment steps applied sequentially to the same tryptic digest — each step capturing a different modification class, enriching the corresponding modified peptides for dedicated LC-MS/MS analysis. This strategy maximizes depth for each individual modification type while maintaining the sample identity across modification classes, enabling direct cross-PTM comparison from the same biological sample.
Standard panel configurations:
- Core signaling panel: Phospho (IMAC) + Ubiquitylation (diGLY IP) — the most commonly requested combination for drug MoA and signal transduction studies.
- Epigenetic panel: Phospho + Acetyl (pan-Ac-Lys antibody) + Methyl (pan-me-Lys/Arg antibody) — for chromatin biology and histone modification research.
- Metabolic acyl panel: Acetyl + Crotonyl + Succinyl + Lactyl — for metabolic reprogramming and Warburg effect research.
- Multi-PTM redox panel: Phospho + Cysteine redox (differential alkylation) + Acetyl — for oxidative stress and redox signaling studies (see also Redox PTM Proteomics).
- Custom panel: any combination of 2–4 modification types with compatible enrichment chemistries — discussed during project consultation.
Depth per PTM type: comparable to dedicated single-PTM workflows — 5,000–20,000+ phosphosites, 3,000–10,000 acetylation sites, 5,000–15,000 K-ε-GG ubiquitylation sites — from the same starting digest.
Strategy 3 — Unenriched Deep Proteome + PTM Profiling
High-depth DIA or TMT-based total proteome analysis from an unenriched digest, combined with variable modification database searching that includes all major PTM types as variable modifications. This approach identifies high-stoichiometry modifications (constitutive phosphorylation on abundant proteins, N-terminal acetylation, abundant glycosylation, histone marks) directly from the total proteome data without a dedicated enrichment step.
Advantage: provides both protein abundance and modification data from the same experiment — no additional sample is consumed for modification analysis. Particularly valuable for low-input samples (single cells, rare biopsies, microdissected tissues) where sample quantity does not permit sequential enrichment.
Limitation: detection is limited to modifications present at ≥1–5% occupancy on proteins that are identified at sufficient depth in the unenriched proteome. Low-stoichiometry modifications on low-abundance proteins will not be detected without enrichment.
Instruments: Bruker timsTOF Pro (diaPASEF) or Thermo Orbitrap Fusion Lumos for maximum unenriched proteome depth; EThcD fragmentation for labile modifications; variable modification search in Spectronaut (DIA) or MaxQuant (DDA).
Global PTM Profiling Service Options
Standard Global PTM Profiling
Open-search discovery analysis of a tryptic digest using MSFragger or MetaMorpheus G-PTM-D — identifying all detectable modification types and their sites in the unenriched sample. Includes PTM-Shepherd post-processing for modification type summarization, site annotation against PhosphoSitePlus and UniMod, and a comprehensive modification landscape report. Suitable as a first-pass survey experiment preceding targeted single-PTM enrichment studies.
Input: ≥100 μg total protein (cells or tissue); ≥200 μL plasma/serum (depleted recommended).
Deliverables: raw data, modified peptide identification table (all modification types), modification type distribution summary, site annotation against PTM databases, modification co-occurrence matrix, bioinformatics figures, methods text, project report.
Multi-PTM Sequential Enrichment Profiling
Two to four modification-specific enrichments applied sequentially to the same protein digest — generating deep site-level data for each modification type from the same biological sample. Includes integrated cross-modification analysis: proteins regulated in two or more modification types are identified, co-modification correlation is calculated, and pathway-level network analysis annotates crosstalk between modification pairs. The most comprehensive global PTM dataset available from a single CRO engagement.
Input: ≥3–10 mg total protein per sample (varies by number of enrichment steps).
Deliverables: all single-PTM deliverables for each modification type covered, plus: cross-modification correlation matrix, co-regulated protein list, PTM crosstalk network visualization, integrated pathway enrichment across modification types, combined publication-ready figure set.
Comparative Global PTM Profiling
Global PTM Profiling applied across two or more biological conditions — treatment vs. control, disease vs. normal, or time-course samples — enabling identification of which modification types and specific sites change between conditions. Uses label-free, TMT, or SILAC quantification strategies depending on sample type and experimental design. Generates a differential modification landscape: modification types with overall regulation, proteins with regulation in multiple PTM types, and pathway-level analysis of regulated modification networks.
Best for: initial characterization of the PTM response to a drug, genetic perturbation, or disease state — before committing to single-PTM deep quantification workflows.
Input: ≥3–5 mg per sample (LFQ), ≥2 mg (TMT), ≥5 mg (SILAC); minimum 3 biological replicates per condition.
Low-Input Global PTM Profiling
Specialized global PTM Profiling workflow for rare or limited samples — FFPE tissue sections, single sorted cell populations (≥50,000 cells), laser-capture microdissected material, or precious clinical biospecimens where standard input amounts are not available. Uses SP3-based sample preparation, nanoscale LC gradients, and Bruker timsTOF Pro or Orbitrap Fusion Lumos configured for maximum sensitivity at low peptide loads. Open-search + variable modification search from unenriched digest provides breadth-limited but unbiased PTM survey data from sub-microgram protein inputs.
Protein-Specific PTM Landscape Mapping
Global PTM characterization focused on a single protein or protein complex of interest — immunoprecipitated or affinity-purified before digestion. All modification types on the captured protein are identified simultaneously by LC-MS/MS with variable modification search across all Unimod entries, providing a complete modification map of the target protein including unexpected, low-stoichiometry, or cell-state-dependent modifications that no pre-specified modification search would identify.
Applications: characterization of recombinant protein products; modification mapping of a protein of pharmacological interest; IP-MS modification characterization before functional mutagenesis studies.
Global PTM + Total Proteome Integration
Combined global PTM profiling and total proteome quantification from the same sample — providing both protein abundance and modification data in a unified project. The 5% pre-enrichment aliquot retained from each sample is analyzed by DIA or TMT proteomics, and protein abundance ratios are used to normalize modification fold-changes for abundance effects — delivering modification-intrinsic regulation data. Also enables identification of proteins where regulation occurs exclusively at the PTM level with no change in total abundance — a biologically important class of regulatory events invisible to total proteomics alone.
Global PTM Profiling Workflow
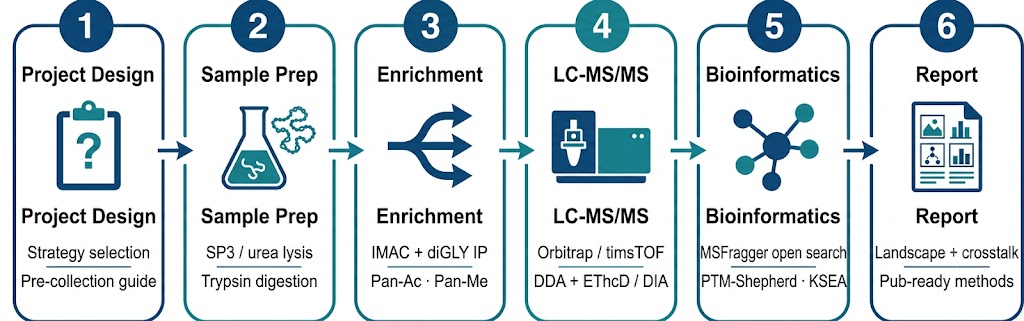
Step 1 — Project Design & Strategy Selection
We review your sample type, biological question, modification types of interest (if any), available input, and publication goals. Based on this, we recommend the optimal profiling strategy — open search, sequential enrichment, or integrated total proteome + PTM — and specify the enrichment panel configuration if applicable. Pre-collection handling requirements are confirmed: phosphatase inhibitors (phospho), NEM (ubiquitylomics), snap-freeze protocols (histone stability). We provide a complete sample collection guide before you collect samples.
Step 2 — Sample Preparation
Protein extraction uses matrix-appropriate lysis buffers — denaturing (8 M urea) for maximum protein solubilization; acid extraction for histone modification studies; non-denaturing for interaction-preserving applications. Reduction (DTT), alkylation (chloroacetamide), and trypsin digestion are performed under standardized conditions validated for each matrix. Peptide yield and missed cleavage rate are measured. A pre-enrichment aliquot (5%) is retained from each sample for total protein abundance quantification. For TMT projects, isobaric labeling and pooling are performed before the enrichment stage.
Step 3 — Enrichment (Strategy-Dependent)
For sequential multi-PTM enrichment: each enrichment step is applied to aliquots from the same digest in series. Order is optimized to maximize recovery: phospho IMAC is applied first (highest recovery from unenriched digest), followed by antibody-based enrichments (diGLY IP, pan-Ac-Lys, etc.), each using the post-IMAC flow-through or fresh aliquots depending on compatibility. Enrichment efficiency is monitored per step using spiked synthetic peptide standards. For open-search profiling: the unenriched digest is analyzed directly — no enrichment required.
Step 4 — LC-MS/MS Acquisition
Acquisition mode and platform are matched to the profiling strategy. For open-search discovery: DDA on Orbitrap Fusion Lumos (high MS/MS scan rate for broad PTM fragmentation coverage) with HCD at 27–30% NCE; EThcD available for labile modifications. For sequential enrichment (deep coverage per PTM): DDA (SILAC/LFQ) or DIA (large cohorts); diaPASEF on Bruker timsTOF Pro for ion-mobility-enhanced depth. For total proteome + PTM integration: DIA on Q Exactive HF-X or timsTOF Pro for the proteome fraction, DDA for the enriched PTM fractions. Instrument QC includes iRT injection, mass accuracy check, and spray stability verification before each run.
Step 5 — Integrated Bioinformatics Analysis
Open-search data is processed in FragPipe (MSFragger open search + PTM-Shepherd) and/or MetaMorpheus G-PTM-D with UniMod-curated and novel modification libraries; 1% FDR at PSM and protein level; modification localization by MSFragger site scoring. Enrichment-based data is processed in MaxQuant (SILAC/LFQ) or Proteome Discoverer (TMT) with modification-specific variable modifications, ptmRS/phosphoRS site localization. For integrated multi-PTM projects: cross-modification analysis identifies proteins present in two or more modification datasets; Pearson correlation between modification-type fold-changes is calculated per protein; PTM crosstalk networks are constructed using STRING PPI data.
Step 6 — Publication-Ready Report Generation
All analysis outputs are compiled into a structured deliverable package organized for direct use in manuscript preparation. The report includes: a modification landscape overview figure (pie chart of modification type distribution, protein-level modification frequency histogram), site confidence summary (localization probability distribution), differential regulation results per modification type (volcano plots, heatmaps), cross-modification co-regulation analysis, GO/KEGG/Reactome pathway enrichment figures, PTM crosstalk network diagram, and a complete Materials and Methods section written at publication standard — covering sample preparation, enrichment, instrument parameters, database search, quantification, and statistical analysis. All figures are provided as editable vector files (PDF or SVG) and high-resolution PNG for direct manuscript use.
Publication-Ready Deliverables — What You Receive

Every Global PTM Profiling project is designed and delivered to support direct manuscript preparation. Our deliverable package is structured around what a peer-reviewed publication requires — not just a data file dump.
Raw & Processed Data Files
- Raw LC-MS/MS data files in Thermo .raw or Bruker .d format
- FragPipe/MaxQuant/Proteome Discoverer project files with all search parameters
- Modified peptide identification table: sequence, charge state, modified residue(s), modification type, mass shift, localization probability score, spectral FDR, protein accession
- Site-level quantification matrix (if quantitative): normalized intensities or ratios across all samples and replicates
- Protein-level abundance matrix from pre-enrichment aliquot (for integrated projects)
Modification Landscape Figures
- Modification type distribution: pie or bar chart showing the number of identified sites per modification class (phosphorylation, acetylation, methylation, ubiquitylation, acylation, etc.)
- Modified amino acid frequency: bar chart of modification frequency per residue type (Ser, Thr, Tyr, Lys, Arg, Cys, Met, Asn)
- Protein-level modification frequency histogram: number of distinct modification types per protein, identifying heavily modified regulatory proteins
- Site localization probability distribution: violin or histogram confirming high-confidence site assignments
- Co-modification heatmap: proteins carrying multiple modification types, with modification type as rows and proteins as columns
Differential Analysis Results (Comparative Projects)
- Volcano plots per modification type: log2 fold-change vs. −log10(adjusted p-value), significantly regulated sites highlighted, top sites annotated
- Hierarchical clustering heatmap of significantly regulated sites across conditions
- Modification-type regulation summary: number of up/down-regulated sites per PTM class per comparison
- Overlap Venn diagram: proteins with regulated sites in two or more modification types
- Cross-modification correlation scatter plot: modification fold-changes plotted against each other for shared regulated proteins
Pathway & Network Analysis
- GO biological process, molecular function, and cellular component enrichment — bubble plots for each modification type
- KEGG and Reactome pathway enrichment — top 15 pathways per modification type, with regulated proteins mapped
- Kinase-substrate enrichment analysis (KSEA) for phospho data — kinase activity inference from regulated phosphosite substrates
- PTM crosstalk network: STRING PPI network of proteins regulated in multiple modification types, with edge color coding modification pair co-regulation
- Modification motif analysis: sequence logos of modification site flanking sequences, colored by upstream kinase or writer enzyme family
Publication-Ready Methods Section
A complete, accurate, publication-standard Materials and Methods section covering every aspect of the experiment — written at the level expected by high-impact proteomics journals (Mol Cell Proteomics, J Proteome Res, Nat Commun, Cell Reports). Covers:
- Sample preparation (lysis, reduction, alkylation, digestion, optional fractionation)
- Enrichment protocols (resin type, batch size, wash conditions, elution)
- LC-MS/MS parameters (column, gradient, instrument model, acquisition mode, resolution, AGC, fill time)
- Database search parameters (software version, database, enzyme, variable modifications, FDR thresholds, site localization algorithm)
- Quantification and normalization approach
- Statistical analysis (test type, multiple testing correction, significance thresholds)
- Bioinformatics tools (software versions, databases, visualization tools)
Comprehensive Project Report
- Executive summary of key findings per modification type
- QC metrics: peptide identification depth, FDR, enrichment efficiency, inter-replicate Pearson correlation, CV distribution
- Interpretation of modification landscape in the context of the submitted biological question
- Recommendations for downstream follow-up: priority modification types for deeper quantitative profiling, priority sites for PRM verification, suggested crosstalk relationships for functional validation
- All figures provided as editable PDF/SVG + high-resolution PNG (300 dpi minimum), formatted for direct manuscript insertion or journal figure preparation
Case Study — Integrative Multi-PTM Proteomics Reveals Coordinated Redox, Phosphorylation, and Acetylation Regulation in Cytokine-Treated Pancreatic β-Cells
Reference: Gluth A, Li X, Gritsenko MA, et al. Integrative multi-PTM proteomics reveals dynamic global, redox, phosphorylation, and acetylation regulation in cytokine-treated pancreatic beta cells. Mol Cell Proteomics. 2024;23(12):100881. DOI: 10.1016/j.mcpro.2024.100881 (CC BY 4.0, PMC11700301)
Background & Scientific Question
Pancreatic β-cell dysfunction driven by cytokine-mediated inflammation is a central mechanism in type 1 diabetes. The regulatory response to cytokine stress in β-cells is known to involve multiple PTM layers — JAK-STAT and NF-κB signaling (phosphorylation), epigenetic reprogramming (acetylation), and oxidative stress-driven protein modification (cysteine thiol oxidation). However, these layers had predominantly been studied in isolation: separate experiments, separate samples, and separate analytical workflows — making it impossible to identify regulatory crosstalk between modification types or to determine which proteins are co-regulated at the PTM level across multiple modification classes simultaneously.
Gluth et al. from PNNL asked: can a single integrated sample preparation workflow quantify protein abundance, cysteine thiol oxidation, phosphorylation, and acetylation from the same samples in the same time-course experiment — and what co-regulatory relationships between these PTM types emerge when you do?
Methods
The study developed an integrative SP3-based workflow that applies four analytical dimensions to the same sample: (1) total protein abundance by DIA proteomics from the unenriched fraction; (2) cysteine thiol oxidation by differential alkylation with iodoacetamide (reduced cysteines) and MMTS (oxidized cysteines) followed by enrichment; (3) phosphorylation by IMAC enrichment; and (4) acetylation by pan-acetyllysine antibody enrichment.
MIN6 mouse β-cell cultures were treated with a cytokine mixture (IL-1β, IFN-γ, TNF-α) across a six-point time course (0, 1, 3, 6, 12, 24 h), with four biological replicates per time point. TMT11-plex labeling enabled all 11 time point–replicate combinations to be compared within a single LC-MS/MS run per modification type. The integrated dataset comprised protein abundance, cysteine oxidation, phosphorylation, and acetylation measurements from the same 44 samples.
Results Overview
The integrated multi-PTM workflow identified thousands of regulated events across all four analytical dimensions. Global protein abundance analysis revealed a rapid response consistent with JAK-STAT and NF-κB pathway activation within 1–3 h of cytokine treatment. Cysteine thiol oxidation showed a distinct temporal profile — peaking at 6–12 h — consistent with a secondary oxidative stress wave following initial immune activation. Phosphoproteomics identified thousands of regulated phosphosites with temporal profiles mapping to kinase activation sequences in JAK-STAT, MAPK, and NF-κB pathways. Acetylomics revealed time-dependent changes in histone and non-histone protein acetylation linked to transcriptional reprogramming during prolonged cytokine stress.
The key biological insight only accessible through integrated multi-PTM analysis: PARP14, a negative regulator of JAK-STAT signaling, was identified with multiple co-localized PTMs — phosphorylation, acetylation, and cysteine oxidation on the same protein, at different time points — suggesting intraprotein PTM crosstalk as a mechanism for fine-tuning JAK-STAT suppression during the acute-to-chronic inflammatory transition. This finding would have been invisible to any single-PTM study.
Relevance to Our Service
This study is the definitive demonstration of what integrated multi-PTM profiling delivers that single-PTM workflows structurally cannot: the identification of proteins co-regulated across multiple modification types, the temporal co-ordination between modification classes, and the discovery of functional PTM crosstalk relationships on specific regulatory proteins. The PARP14 multi-PTM finding is a prototype of the type of discovery that only emerges when phosphorylation, acetylation, and redox modifications are profiled from the same samples in the same experiment.
Our Multi-PTM Sequential Enrichment Profiling service uses the same conceptual framework — same sample, multiple enrichment steps, integrated analysis — enabling this class of discovery in your biological system. The publication-ready deliverables from our platform include all the figure types required to present integrated multi-PTM data in high-impact journals, including the cross-modification temporal heatmaps, co-regulation scatter plots, and PTM crosstalk network visualizations demonstrated in this paper.
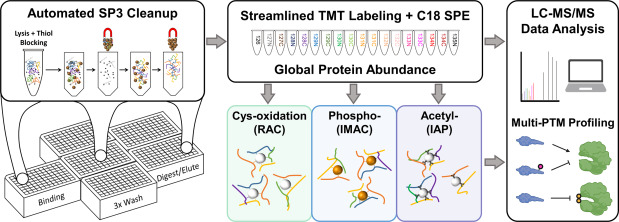
Adapted from Gluth et al. 2024, Mol Cell Proteomics 23:100881, CC BY 4.0, PMC11700301. Integrated multi-PTM temporal profiling in cytokine-treated β-cells — illustrating the type of cross-modification co-regulation analysis delivered as part of our Global PTM Profiling service.
References
- Gluth A, Li X, Gritsenko MA, et al. Integrative multi-PTM proteomics reveals dynamic global, redox, phosphorylation, and acetylation regulation in cytokine-treated pancreatic beta cells. Mol Cell Proteomics. 2024;23(12):100881. doi.org/10.1016/j.mcpro.2024.100881
- Chick JM, Kolippakkam D, Nusinow DP, et al. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat Biotechnol. 2015;33(7):743-749. doi.org/10.1038/nbt.3267
- Solntsev SK, Shortreed MR, Frey BL, Smith LM. Enhanced global post-translational modification discovery with MetaMorpheus. J Proteome Res. 2018;17(5):1844-1851. doi.org/10.1021/acs.jproteome.7b00873
- Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods. 2017;14(5):513-520. doi.org/10.1038/nmeth.4256
- Krug K, Mertins P, Zhang B, et al. A curated resource for phosphosite-specific signature analysis. Mol Cell Proteomics. 2019;18(8):1457-1474. doi.org/10.1074/mcp.TIR118.000943
FAQs — Global PTM Profiling
What is the difference between Global PTM Profiling and a standard phosphoproteomics or ubiquitylomics experiment?
A standard phosphoproteomics or ubiquitylomics experiment applies a single modification-specific enrichment (IMAC for phospho, diGLY IP for ubiquitylation) and detects only that modification class — by design. It maximizes depth for the targeted modification but is structurally blind to all other PTMs present in the sample. Global PTM Profiling is designed to characterize the complete modification landscape without this tunnel vision: either through open-search analysis of an unenriched sample (detecting all modifications at sufficient stoichiometry without enrichment) or through sequential multi-PTM enrichment (applying two to four enrichment steps to the same digest, capturing each modification class in turn). The result is a multi-modification dataset from a single sample, enabling co-modification analysis and cross-PTM comparison that single-PTM experiments cannot provide. If you already know which modification class you want to study at maximum depth, a dedicated single-PTM workflow (phosphoproteomics, ubiquitylomics, acetylomics) will deliver greater depth than global profiling. If you want to know which PTMs are present and regulated before committing to a single-modification workflow — or if you want cross-modification co-regulation data from the same samples — Global PTM Profiling is the right experiment.
How many modification types and sites can I expect to identify from Global PTM Profiling?
Coverage depends strongly on which profiling strategy is used. Open-search analysis of an unenriched sample typically identifies 20–30 modification types and 5,000–15,000 unique modified sites per experiment from well-characterized cell line proteomes, with the most abundant being phosphorylation (often 40–60% of modified sites in actively signaling cells), N-terminal acetylation, methionine oxidation, and deamidation. For less-abundant modification types (Lys acetylation, Lys methylation, ubiquitylation), site numbers from open-search without enrichment are typically 100–1,000 — much lower than dedicated enrichment workflows but sufficient to identify the most abundant events. Sequential multi-PTM enrichment adds depth dramatically: phospho IMAC + diGLY IP + pan-Ac antibody together from the same digest can deliver 10,000–25,000 phosphosites, 3,000–10,000 K-ε-GG sites, and 3,000–8,000 acetylation sites — all from the same protein digest. Exact coverage for your sample type and input amount is estimated during project consultation based on your specific biological system.
What does "publication-ready" mean in practice — what figures and text will I receive?
Publication-ready means the deliverable package is structured specifically for manuscript preparation, not just data archiving. Concretely, you receive: (1) a Materials and Methods section written at the level expected by Mol Cell Proteomics, J Proteome Res, Nat Commun, or equivalent journals — covering sample preparation, enrichment protocols, instrument parameters, database search settings, quantification, normalization, and statistical analysis, at the level of detail required for methods reproducibility; (2) a set of figures produced at 300 dpi minimum in both editable vector format (PDF/SVG) and high-resolution PNG — ready for direct insertion into a manuscript or figure panel. These include modification landscape overviews, volcano plots, heatmaps, KSEA plots, pathway enrichment bubble plots, PPI/crosstalk networks, and modification motif sequence logos; (3) a results summary narrative describing the key findings in language suitable for a Results section draft — not a database readout, but an interpreted synthesis of what the data shows, organized around the biological question you provided at project initiation. You are not expected to reformat any output for submission — our deliverables are pre-formatted for journal standards. If a reviewer requests specific additional analyses or reanalysis, we offer post-delivery support for figures requested during peer review.
Can open-search discovery detect modifications that no one has described before?
Yes — with important caveats. Open-search algorithms (MSFragger, MetaMorpheus G-PTM-D) identify mass shifts on peptides that do not match any standard modification in the closed variable modification list. These can represent truly novel modifications, but they can also represent known modifications not included in the search database, chemical artifacts from sample preparation (e.g., missed alkylation, in-source fragmentation), or ambiguous mass assignments where multiple modification possibilities have the same nominal mass. Our bioinformatics workflow includes PTM-Shepherd post-processing, which performs residue-specificity analysis (does the mass shift localize preferentially to one amino acid type?) and cross-references against the full UniMod database to classify each mass shift as: (a) a known modification not previously included in the closed search, (b) a known artifact, or (c) a potentially novel modification requiring orthogonal confirmation. We report all three categories clearly in the deliverable, with confidence tiers. For novel modification candidates that pass our specificity and FDR criteria, we recommend orthogonal validation — typically chemical derivatization, specific antibody confirmation, or genetic perturbation of the modification enzyme — before reporting them as novel discoveries in a manuscript.
How does Global PTM Profiling feed into downstream targeted experiments?
Global PTM Profiling is explicitly designed as the discovery phase of a structured PTM research pipeline. The site list it generates — prioritized by modification type, localization probability, protein biological relevance, and (in comparative projects) differential regulation — directly informs the design of downstream experiments: (1) for deeper quantification of priority modification types, PTM Quantitative Analysis with DIA, SILAC, or TMT provides site-level fold-change data with statistical rigor across the full regulated site list; (2) for validation of the top 20–100 priority sites with orthogonal, high-confidence quantification, PRM PTM Site Verification confirms discovery findings in independent replicates with defined per-site analytical performance; (3) for absolute quantification of the highest-priority sites (pharmacodynamic biomarkers, regulatory stoichiometry measurements), Modified Peptide Absolute Quantification establishes molar concentrations with calibration curves and defined LOD/LOQ. The global profiling → quantification → verification → absolute quantification pipeline is the complete, publication-grade PTM research workflow — we support all phases within a single coordinated project or as sequential engagements.