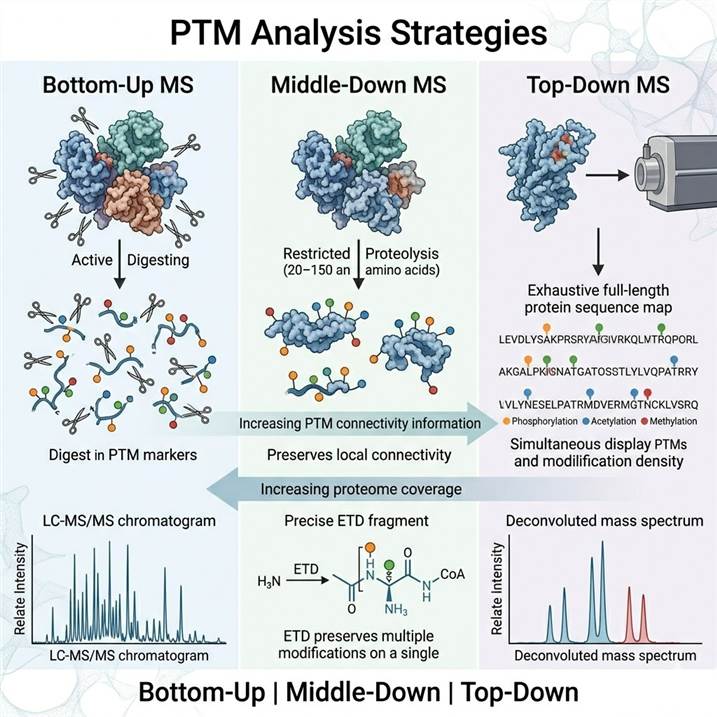
PTM Analysis Strategies: Choosing the Right MS-Based Approach
The choice of PTM analysis strategy fundamentally determines the depth, confidence, and biological relevance of the resulting modification data. Each strategy operates at a different protein structural level — peptides (bottom-up), larger polypeptide fragments (middle-down), or intact proteoforms (top-down) — and each delivers complementary information about PTM identity, site localization, stoichiometry, and combinatorial modification patterns. The selection depends on multiple interdependent factors including the PTM type and its stability during sample processing, the required site localization confidence, the need to preserve modification co-occurrence information, sample quantity constraints, and the throughput demands of the study.
Our MS-Based PTM Analysis platform encompasses all three strategies, providing access to the full methodological toolkit for PTM characterization across diverse PTM types and research contexts. The following sections detail each approach, describing the core workflow, analytical strengths and limitations, and the specific PTM research scenarios for which each strategy is best suited.
Bottom-Up MS: The Workhorse of PTM Proteomics
Bottom-up proteomics (also termed shotgun proteomics) is the most widely adopted MS strategy for PTM analysis. In this approach, proteins are enzymatically digested (typically with trypsin) into peptides before LC-MS/MS analysis, with PTM sites identified through database searching of the resulting fragmentation spectra. The method's compatibility with high-resolution chromatographic separation and its ability to analyze complex peptide mixtures make it the method of choice for deep PTM discovery across the proteome.
Core Workflow
- Protein extraction and digestion: Proteins are extracted, reduced, alkylated, and digested with sequence-specific proteases (trypsin, Lys-C, Glu-C, or combinations) to generate peptides averaging 7–25 amino acids in length
- PTM-specific enrichment: Modified peptides are enriched using antibody-based immunoaffinity (e.g., anti-phosphotyrosine, anti-acetyllysine), chemical capture (e.g., biotin-switch for S-nitrosylation, hydrazide chemistry for glycopeptides), or affinity chromatography (IMAC/TiO₂ for phosphopeptides, HILIC for glycopeptides) before LC-MS/MS analysis
- LC-MS/MS acquisition: Enriched peptide mixtures are separated by nano-flow reversed-phase liquid chromatography and analyzed by high-resolution mass spectrometry using data-dependent acquisition (DDA), data-independent acquisition (DIA), or targeted PRM/ MRM methods
- Database searching and PTM localization: MS/MS spectra are searched against protein sequence databases using search engines (MaxQuant, Proteome Discoverer, MSFragger) that consider PTMs as variable modifications, with site localization scored by localization probability algorithms
- Quantification: PTM abundance is quantified through label-free (XIC alignment), isotopic labeling (SILAC, TMT, iTRAQ), or targeted (PRM/MRM) approaches relative to unmodified peptides or internal standards
Strengths and Best Applications
Bottom-up MS offers the deepest proteome coverage of any PTM analysis strategy, routinely identifying thousands of modification sites from complex biological samples. It is the method of choice for global PTM discovery, quantitative PTM profiling across conditions, and targeted validation of specific modification sites. The approach is compatible with all major PTM enrichment strategies and benefits from a mature bioinformatics ecosystem with well-established workflows for PTM identification, localization, and quantification.
Limitations
The principal limitation of bottom-up MS is the loss of connectivity between modification sites. Because proteins are digested into peptides, information about co-occurring modifications on the same protein molecule — combinatorial PTM patterns, modification crosstalk at proximal sites, and the distribution of PTM isoforms (proteoforms) — is lost. Bottom-Up MS-Based PTM Analysis provides full methodological details for this widely adopted approach.
Middle-Down MS: Bridging the Resolution Gap
Middle-down proteomics occupies an intermediate position between bottom-up and top-down approaches. Proteins are digested with restricted proteolysis using enzymes such as Asp-N, Glu-C, or limited Lys-C digestion to generate larger peptides (typically 3–15 kDa, corresponding to 20–150 amino acids). These larger polypeptides retain more sequence context than bottom-up peptides while remaining amenable to LC-MS/MS analysis with conventional reversed-phase chromatography and collision-based fragmentation.
Core Workflow
- Restricted proteolysis: Proteins are digested under controlled conditions using proteases with restricted cleavage specificity (Asp-N, Glu-C, or limited Lys-C) to generate larger polypeptide fragments that span multiple PTM sites
- Size-based fractionation: Digested polypeptides are fractionated by size-exclusion chromatography or SDS-PAGE to enrich the middle-range (3–15 kDa) fraction, removing both undigested proteins and very small peptides
- LC-MS/MS acquisition: Middle-down polypeptides are separated by C4 or C8 reversed-phase chromatography (wider pores for larger polypeptides) and analyzed on high-resolution mass spectrometers using electron transfer dissociation (ETD) or electron capture dissociation (ECD) in addition to HCD
- Database searching with extended variable modifications: MS/MS spectra are searched against sequence databases with consideration of multiple co-occurring PTMs on the same polypeptide, using specialized search tools such as MASCOT TD, ProSight Lite, or pTop
Strengths and Best Applications
Middle-down MS preserves connectivity between modification sites on the same polypeptide, enabling the identification of co-occurring modifications and providing improved sequence coverage compared to bottom-up approaches. The strategy is particularly valuable for characterizing combinatorial PTM patterns on histone tails, N-terminal modification analysis, and studying PTM crosstalk on multi-domain proteins where modification proximity influences function. It represents an effective compromise between the depth of bottom-up and the single-protein resolution of top-down approaches.
Limitations
Middle-down MS requires optimization of digestion conditions to generate the desired polypeptide size range, which can be less reproducible than the complete digestion used in bottom-up workflows. The larger polypeptide size reduces chromatographic resolution and increases spectral complexity, requiring higher-performance mass spectrometers and ETD/ECD fragmentation to achieve comprehensive sequence coverage. Database searching is computationally more demanding due to the expanded search space from co-occurring modifications. Middle-Down MS-Based PTM Analysis provides comprehensive method details and application guidance.
Top-Down MS: Intact Proteoform Characterization
Top-down proteomics analyzes intact proteins without prior digestion, preserving the complete protein sequence and all associated post-translational modifications within a single molecular measurement. This approach provides the most comprehensive view of PTM combinations on individual protein molecules — the proteoform — enabling direct determination of modification co-occurrence, PTM ordering along the sequence, and the distribution of modification patterns across different proteoforms of the same protein.
Core Workflow
- Intact protein purification and fractionation: Proteins are extracted under native or denaturing conditions and fractionated at the protein level using ion exchange chromatography, size-exclusion chromatography, or capillary electrophoresis to reduce sample complexity before MS analysis
- High-resolution MS acquisition of intact protein ions: Intact proteins are introduced into the mass spectrometer via electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI), with high-resolution Orbitrap or FT-ICR instruments (≥120,000 resolution) required to resolve the charge state envelopes and isotopically resolve smaller proteoforms
- Gas-phase fragmentation: Intact protein ions are fragmented using electron transfer dissociation (ETD), ultraviolet photodissociation (UVPD), or higher-energy collisional dissociation (HCD), with ETD and UVPD preferred for PTM localization as they preserve labile modifications during backbone cleavage
- Proteoform identification and PTM mapping: Fragmentation spectra are matched to protein sequences using specialized top-down search tools (ProSight, TopPIC, TDPortal) that consider the intact mass as a filter and map fragment ions to determine PTM identities and positions along the full-length sequence
Strengths and Best Applications
Top-down MS is the only strategy that provides a complete view of the proteoform — the exact combination of all PTMs present on a single protein molecule at a given time. This capability is essential for characterizing combinatorial histone modifications (the "histone code"), mapping PTM patterns on therapeutic antibodies and biopharmaceuticals, determining the order of modification events during protein maturation, and studying PTM crosstalk where modifications at distant sites influence each other's function. Top-down analysis also achieves 100% sequence coverage for smaller proteins, eliminating the "sequence coverage gap" inherent in bottom-up approaches.
Limitations
Top-down MS is technically demanding and currently limited to lower throughput than bottom-up approaches. The complexity of intact protein mixtures challenges separation and detection, with current practical limits of several hundred to a few thousand identified proteoforms per experiment — orders of magnitude fewer than bottom-up PTM site identifications. The approach requires high-resolution mass spectrometers and specialized fragmentation methods, and computational tools for proteoform identification continue to evolve. Despite these limitations, top-down MS provides unique information about PTM combinations that cannot be obtained by any other method. Top-Down MS-Based PTM Analysis provides detailed methodology and application information.
Strategy Comparison and Selection Guide
The table below provides a direct comparison of the three PTM analysis strategies across key performance parameters, followed by a selection framework to guide method choice based on research objectives.
| Parameter |
Bottom-Up MS |
Middle-Down MS |
Top-Down MS |
| Analyte size |
Peptides (7–25 aa) |
Polypeptides (20–150 aa) |
Intact proteins (full length) |
| Proteome coverage |
Deep (thousands of sites) |
Moderate (hundreds of sites) |
Limited (dozens to hundreds of proteoforms) |
| PTM site localization |
High (localization probability scoring) |
High (extended sequence context) |
Complete (full sequence coverage) |
| Combinatorial PTM analysis |
Not possible (PTM connectivity lost) |
Partial (within polypeptide fragments) |
Complete (full proteoform resolution) |
| Throughput |
High (multiplexed DIA/TMT) |
Moderate |
Low to moderate |
| Sample quantity required |
Low (µg level for enriched PTMs) |
Moderate (10–100 µg) |
Higher (50–500 µg per fraction) |
| Fragmentation method |
HCD (primary), ETD, CID |
ETD/ECD, HCD |
ETD, UVPD, HCD |
| Best suited for |
Global PTM discovery, quantitative PTM profiling, targeted site validation |
Histone PTM combinations, multi-PTM polypeptide analysis, N-terminal modifications |
Proteoform characterization, combinatorial PTM mapping, biopharma QC |
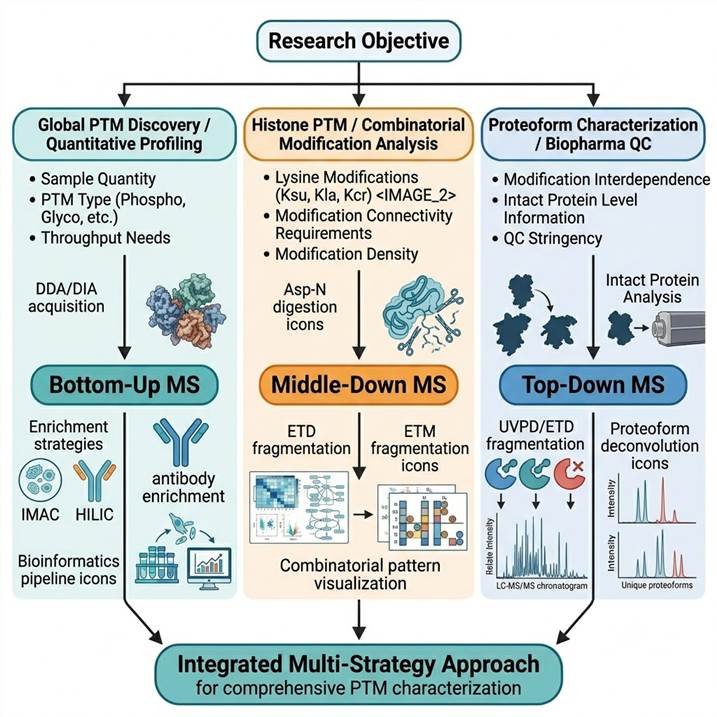
Selection Framework: Matching Strategy to Research Objective
The following decision framework provides rapid guidance for strategy selection based on common PTM research objectives:
- Global PTM discovery and quantitative profiling: Bottom-up MS is the clear first choice. When the goal is to identify as many modification sites as possible and quantify their regulation across conditions — for phosphorylation, acetylation, glycosylation, ubiquitination, or any modification type — bottom-up MS with enrichment delivers the deepest coverage and highest throughput
- Histone PTM and combinatorial modification analysis: Middle-down or top-down MS should be considered. Histone modifications occur in combinations that encode functional information (the "histone code"), and bottom-up MS loses the connectivity between modifications on the same histone tail. Middle-down MS with Asp-N digestion (generating the ~50 amino acid N-terminal tail) preserves combinatorial patterns, while top-down MS provides the complete histone proteoform including both tail and globular domain modifications
- Therapeutic protein and biopharmaceutical PTM characterization: A multi-strategy approach is recommended. Bottom-up MS provides comprehensive glycosylation site mapping and quantification, middle-down MS characterizes glycoform distributions on individual proteolytic fragments, and top-down MS provides intact mass analysis of the complete antibody or fusion protein with all modifications resolved
- Novel PTM discovery and open-search analysis: Bottom-up MS with open-search capabilities (using MSFragger, pFind, or similar) enables unbiased detection of unexpected or novel modifications by searching with wide precursor mass tolerances, making it the method of choice for discovering previously uncharacterized PTMs
- PTM crosstalk and multi-modification coordination: Middle-down or top-down MS provides the connectivity information needed to determine whether two modifications co-occur on the same protein molecule, their relative positions, and potential functional interplay
Related Services
Our PTM analysis platform provides dedicated service pages for each MS strategy, PTM enrichment approach, and bioinformatics solution, enabling researchers to select the specific service modules that match their analytical requirements.
- Pan PTM Proteomics — Comprehensive multi-modification profiling across diverse PTM classes for systems-level PTM landscape characterization
- PTM Proteoform Mapping — Detailed characterization of combinatorial PTM patterns on individual protein proteoforms across all modification types
- Open-Search PTM Discovery — Unbiased open-search approach for detecting unexpected or novel modifications using wide precursor mass tolerance searching
- Antibody-Based Immunoaffinity Precipitation — Targeted enrichment of modified proteins and peptides using modification-specific antibodies across all PTM classes
- Modified Peptide Enrichment Services — Specialized enrichment strategies for low-abundance PTM peptides across all modification classes
- PTM Services — Complete PTM analysis service portfolio covering discovery, quantification, and bioinformatics
- Global PTM Profiling — Broad multi-PTM discovery analysis across diverse modification types from a single biological sample
- PTM Bioinformatics Analysis — Advanced bioinformatics for PTM data integration, functional annotation, and multi-strategy data interpretation
- PTM Crosstalk Analysis — Multi-modification coordination and crosstalk analysis leveraging connectivity data from middle-down and top-down strategies
- PTM in Biological Research — Application-focused PTM analysis solutions for specific biological research contexts
Frequently Asked Questions
What is the difference between bottom-up, middle-down, and top-down MS strategies for PTM analysis?
Bottom-up MS digests proteins into short peptides (7–25 amino acids) before analysis, providing the deepest proteome coverage and highest throughput for PTM site identification. Middle-down MS uses restricted proteolysis to generate larger polypeptides (20–150 amino acids), preserving connectivity between modification sites on the same polypeptide. Top-down MS analyzes intact proteins without digestion, providing complete characterization of all modifications on a single proteoform. The choice between strategies represents a trade-off between coverage depth, throughput, and the retention of combinatorial modification information.
Which MS strategy is best for global PTM discovery?
Bottom-up MS is the method of choice for global PTM discovery, routinely identifying thousands of modification sites from complex biological samples. The approach benefits from mature enrichment strategies, well-established LC-MS/MS workflows, and comprehensive bioinformatics tools for PTM identification and site localization. For unbiased discovery of unexpected or novel modifications, bottom-up MS with open-search capabilities enables detection of modifications without specifying them a priori in the database search.
When should I use middle-down or top-down MS instead of bottom-up?
Middle-down and top-down MS are recommended when information about co-occurring modifications on the same protein molecule is essential — for combinatorial histone modification analysis, characterization of PTM crosstalk on multi-domain proteins, determining modification order, and proteoform-level analysis of therapeutic proteins. Top-down MS is uniquely capable of resolving the full proteoform distribution, revealing how many different modification states exist for a given protein and their relative abundances.
Can I combine multiple strategies in a single study?
Yes — multi-strategy approaches are increasingly used to obtain complementary information. A common design uses bottom-up MS for deep PTM discovery and quantitative profiling across conditions, followed by middle-down or top-down MS for detailed characterization of combinatorial modification patterns on specific proteins of interest. This integrated approach combines the discovery power of bottom-up proteomics with the connectivity information provided by middle-down and top-down methods. Our platform supports all three strategies and can design multi-strategy workflows tailored to your research objectives.
How much sample is needed for each strategy?
Bottom-up MS with PTM enrichment typically requires the least starting material (µg-level protein for targeted PTM analysis to low mg-level for deep global PTM profiling). Middle-down MS requires moderate sample amounts (10–100 µg protein). Top-down MS typically requires larger amounts (50–500 µg per fraction), as intact protein analysis is less sensitive than peptide-level analysis and sample losses during protein-level fractionation are more significant. The specific amount depends on the PTM type, enrichment efficiency, and the required depth of coverage.
What fragmentation methods are used for PTM localization in each strategy?
Bottom-up MS primarily uses higher-energy collisional dissociation (HCD), which provides efficient peptide backbone fragmentation and is compatible with fast scanning required for DIA and multiplexed TMT workflows. Middle-down and top-down MS benefit from electron transfer dissociation (ETD) and ultraviolet photodissociation (UVPD), which preserve labile PTMs during backbone fragmentation — critical for accurate localization of phosphorylation, glycosylation, and other modification types that can be lost during collisional activation. The choice of fragmentation method is tailored to the specific PTM type and analytical objective.
How do I choose the right strategy for my research project?
Strategy selection should be guided by three primary factors: (1) whether combinatorial modification information is required — if yes, middle-down or top-down MS is recommended; (2) the depth of coverage needed — bottom-up MS provides the broadest coverage and is preferred for global discovery; and (3) sample quantity and complexity constraints. Our PTM scientists provide free consultation to help match the strategy to your specific research objectives, sample characteristics, and desired data outputs. Contact us to discuss your project requirements.
For research use only. Not for use in diagnostic procedures.