Why Absolute Quantification of Modified Peptides Matters
Most PTM studies report relative quantification — the fold-increase or decrease of a modified peptide between conditions. While useful for identifying candidates, relative quantification has fundamental limitations when the goal is to understand mechanism, build predictive models, or compare across independent experiments.
Absolute quantification addresses these gaps:
- Determines site occupancy directly — The fraction of a protein population that carries a specific modification at a given site (stoichiometry) can only be determined through absolute measurement of both the modified and unmodified forms. Knowing that a kinase substrate is "2-fold upregulated" is less informative than knowing its phosphorylation stoichiometry increased from 5% to 45% — the latter reveals that the site transitions from a minor species to a dominant one, a distinction with profound signaling implications.
- Enables cross-experiment and cross-laboratory comparison — Relative ratios are tied to the specific experimental context. Absolute values — expressed in fmol/mg protein, copies/cell, or molar concentration — are universally comparable, making them invaluable for multi-site collaborations, longitudinal studies, and meta-analyses.
- Supports quantitative modeling — Mathematical models of signaling networks, enzyme kinetics, and drug-target engagement require absolute abundances and stoichiometries as input parameters. Relative data alone cannot inform such models.
- Provides definitive biomarker quantification — In clinical proteomics and biomarker development, absolute concentrations are required for assay validation and regulatory compliance. For deeper functional analysis of PTM-modified proteins detected in your study, explore our PTM Bioinformatics Analysis service.
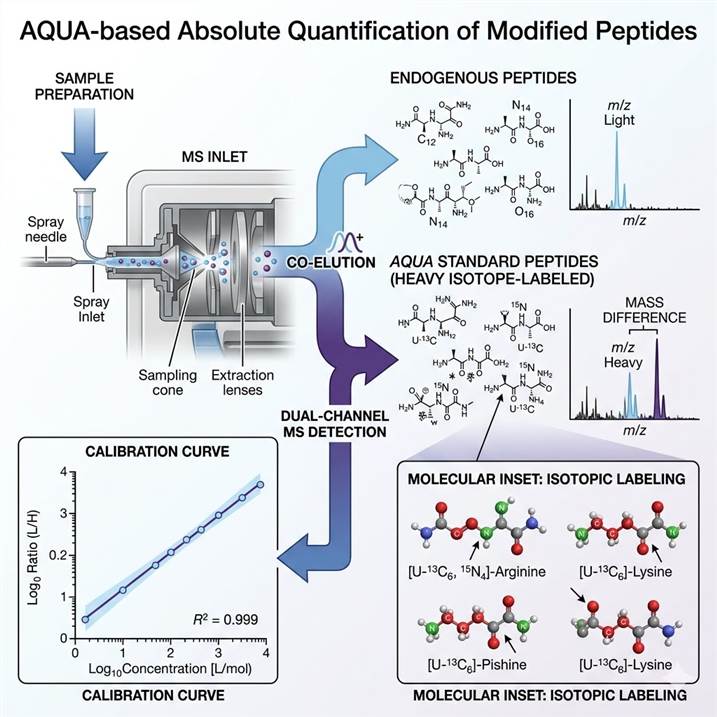
AQUA-Based Strategy for Modified Peptide Absolute Quantification
The core technology underlying our absolute quantification service is the AQUA (Absolute QUAntification) strategy. AQUA uses synthetic peptides labeled with stable heavy isotopes (¹³C, ¹⁵N) as internal standards that are chemically identical to the target endogenous peptide but distinguishable by mass.
How AQUA Enables Absolute Quantification
Target peptide selection: The modified peptide of interest (e.g., a specific phosphopeptide, acetylated peptide, or ubiquitin remnant-containing peptide) is identified through prior discovery experiments or literature review.
AQUA internal standard synthesis: A synthetic equivalent of the target peptide is produced with one or more heavy isotope-labeled amino acids (e.g., [¹³C₆]Arg, [¹³C₆¹⁵N₄]Arg). This AQUA peptide has identical chromatographic and ionization properties to the endogenous peptide but is +6 to +10 Da heavier, making it distinguishable by mass spectrometry.
Standard curve construction: A known amount of AQUA peptide is spiked into each sample. By comparing the MS signal of the endogenous (light) peptide to the known amount of the AQUA (heavy) internal standard, the absolute quantity of the endogenous modified peptide is calculated.
Multiplexed quantification: Multiple AQUA peptides — targeting different modified sites or different modification types — can be multiplexed in a single PRM or SRM acquisition, enabling high-throughput absolute quantification across a panel of targets. For targeted verification of specific PTM sites, our PRM PTM Verification service provides dedicated assay development and validation.
PRM and SRM Acquisition Platforms
We deploy both parallel reaction monitoring (PRM) on high-resolution Orbitrap instruments and selected reaction monitoring (SRM, also called MRM) on triple-quadrupole platforms:
| Parameter |
PRM (Orbitrap) |
SRM/MRM (Triple-Quad) |
| Resolution |
30,000–120,000 |
Unit (Q1/Q3) |
| Selectivity |
Full MS/MS scan |
Transition-specific |
| Multiplexing capacity |
Moderate (15–30 targets) |
High (50–200+ transitions) |
| Sensitivity |
Low-attomole |
Low-attomole |
| Best suited for |
High-specificity, complex matrix |
High-throughput, large panels |
Site Occupancy Determination
When both the modified and unmodified versions of a peptide can be monitored, we calculate site occupancy (stoichiometry) as: Site Occupancy (%) = [Modified Peptide] / ([Modified Peptide] + [Unmodified Peptide]) × 100. This calculation is performed with internal standard correction for both forms, ensuring accurate stoichiometry values even when the modified and unmodified peptides differ in ionization efficiency. For complementary relative quantification workflows, our Label-Free PTM Quantification service provides broad-coverage quantitative PTM profiling without isotopic labeling.
Compatible Sample Types and Material Requirements
Our absolute quantification workflow accepts samples from any biological source and can handle essentially any modification type with an appropriate AQUA standard.
Accepted Sample Types
| Sample Type |
Minimum Requirement |
Notes |
| Cultured cells (mammalian) |
≥1 × 107 cells |
Adherent or suspension; metabolic labeling optional |
| Cultured cells (yeast, bacteria) |
≥5 × 107 cells |
Higher input for lower protein per cell |
| Tissues |
≥10 mg wet weight |
Snap-frozen or RNAlater-stabilized |
| Biofluids (plasma/serum) |
≥100 μL |
High-abundance protein depletion optional |
| Biofluids (CSF, urine, supernatant) |
≥500 μL |
May require concentration step |
| Subcellular fractions |
≥50 μg protein equivalent |
Nuclear, cytoplasmic, membrane, mitochondrial |
| Immunoprecipitated samples |
≥5 μg protein per pull-down |
On-bead digestion supported |
| Purified protein complexes |
≥1 μg of target protein |
Minimal background, highest sensitivity |
Supported Modification Types
| Modification Class |
Examples |
| Phosphorylation |
pSer, pThr, pTyr — single and multi-site stoichiometry |
| Acetylation |
Lysine acetylation, N-terminal acetylation |
| Ubiquitination |
diGly remnant (K-ε-GG), ubiquitin chain linkage analysis |
| Methylation |
Mono-, di-, tri-methylation (Lys, Arg) |
| Oxidation |
Methionine oxidation, cysteine oxidation |
| Acylation |
Lactylation, succinylation, crotonylation, butyrylation |
| Glycosylation |
N-glycosylation, O-GlcNAc site occupancy |
| SUMOylation |
SUMO remnant peptides |
| Custom modifications |
Any modification accessible via AQUA peptide design |
For broader quantitative PTM profiling across many targets, consider our DIA/SWATH PTM Quantification service for data-independent acquisition-based quantification.
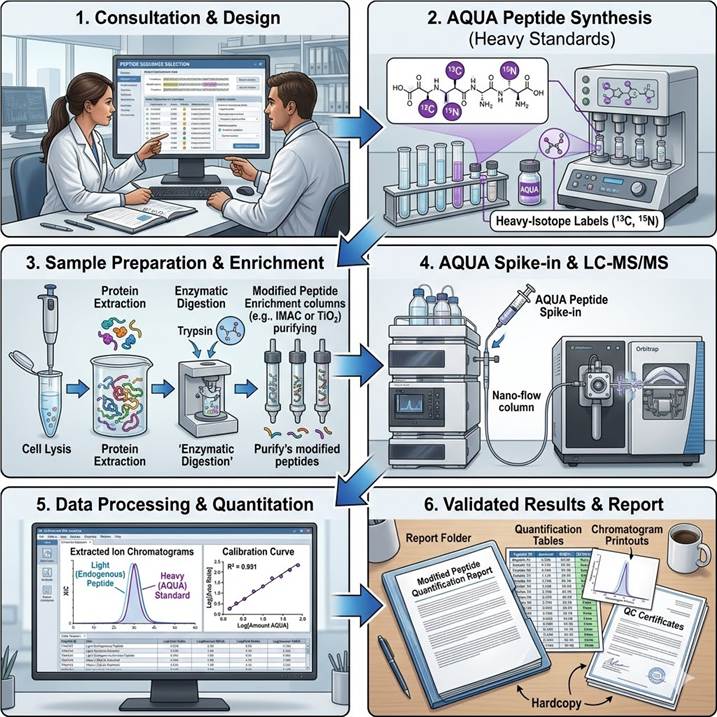
End-to-End Workflow: From Sample to Absolute Quantification Data
Step 1: Consultation and Target Design
We begin with a detailed consultation to define your quantification targets. You provide the modified peptide sequences of interest, modification types, and specific sites. Our team designs AQUA internal standard peptides, selecting optimal heavy isotope labeling positions and verifying sequence uniqueness against the target proteome.
Step 2: AQUA Peptide Synthesis and QC
Stable isotope-labeled AQUA peptides are synthesized with ≥97% isotopic purity. Each batch undergoes QC: accurate mass confirmation by MS, purity assessment by LC-UV, quantitative amino acid analysis for concentration determination, and spike-in recovery testing in the relevant matrix. Typical turnaround: 3–4 weeks.
Step 3: Sample Preparation and Digestion
Samples are processed under standardized conditions. Protein extraction, reduction, alkylation, and digestion (trypsin/Lys-C or alternative proteases) follow established protocols. Where applicable, enrichment of the modified peptide population is performed using validated methods (IMAC, TiO₂, antibody-based enrichment).
Step 4: AQUA Spike-In and LC-MS/MS
Known quantities of AQUA internal standards are spiked into each digested sample prior to LC-MS analysis. Nano-flow reverse-phase chromatography (C18, 75 μm × 25 cm column, 120-min gradient) is coupled to high-resolution Orbitrap or triple-quadrupole mass spectrometry for PRM or SRM acquisition with scheduled retention times.
Step 5: Data Processing and Quantification
Raw MS data are processed using validated quantification pipelines: extraction of endogenous (light) and AQUA (heavy) peptide chromatograms, peak area integration with quality metrics, calculation of absolute quantities via standard curve regression, site occupancy calculation (when applicable), and error propagation across replicates.
Step 6: Deliverables and Review
Raw MS data files, AQUA peptide QC certificates, extracted ion chromatograms for each target, absolute quantification table (fmol/mg, copies/cell, or molar concentration), site occupancy percentages, assay performance metrics (LOD, LOQ, linear range, CV%), and a scientist consultation session for data walk-through.
Why Choose Our Modified Peptide Absolute Quantification Service
AQUA-PRM Expertise Across Modification Classes
We have extensive experience designing, validating, and deploying AQUA-based absolute quantification assays for phosphorylation, acetylation, ubiquitination, methylation, and emerging modification types. Our team has optimized protocols for phosphopeptide stoichiometry, histone mark quantification, and ubiquitin chain linkage analysis — spanning the full diversity of the PTM landscape.
Dual-Platform Flexibility
With access to both high-resolution Orbitrap (PRM) and triple-quadrupole (SRM/MRM) platforms, we select the optimal instrumentation for each project. PRM offers superior specificity for low-abundance modified peptides in complex matrices; SRM provides higher throughput and lower cost-per-target for established panels. Explore our MS-Based PTM Analysis platform for full technical specifications.
Full Assay Validation
Every AQUA assay we deliver includes linearity assessment (R² ≥ 0.99), limit of detection (LOD), lower limit of quantification (LLOQ), intra- and inter-assay precision (CV ≤ 20%), and spike-recovery validation. This rigorous validation ensures your quantification data meets the highest standards for publication and regulatory submission support.
Integrated Site Occupancy Analysis
For projects requiring stoichiometry determination, we simultaneously quantify both modified and unmodified peptide forms using matched AQUA internal standards. This dual-quantification approach eliminates the ionization efficiency bias that confounds label-free stoichiometry estimates.
End-to-End Project Management
From AQUA peptide design through data delivery, a dedicated project scientist manages your workflow, provides regular updates, and ensures timelines are met. Each project concludes with a consultative data review session.
Representative Results: Absolute Quantification Data Outputs
Our absolute quantification data packages deliver far more than raw numbers. Every project includes fully annotated quantification results with quality metrics, enabling immediate assessment of data reliability and biological significance.
Representative data outputs from our absolute quantification pipeline. Left: Extracted ion chromatograms for endogenous and AQUA internal standard peptide pairs. Center: Calibration curve demonstrating linear quantification across 4 orders of magnitude. Right: Site occupancy comparison across treatment conditions.
Key data components included in every deliverable package:
- Extracted ion chromatograms — Overlay of endogenous (light) and AQUA (heavy) peptide signals with co-elution verification for each target
- Calibration curves — Linear regression (R² ≥ 0.99) spanning 3–5 orders of magnitude with LOD and LLOQ marked
- Absolute quantification table — Molar quantities per injection (fmol on-column), normalized to protein input (fmol/mg), and per-cell copy numbers (copies/cell) where applicable
- Site occupancy matrix — Stoichiometry values for each modified site across all samples and conditions
- Assay performance summary — Intra- and inter-assay precision (CV%), spike-recovery accuracy, and measurement uncertainty
Case Study: DM+deP Phosphorylation Stoichiometry Validated by AQUA-SRM Absolute Quantification
In a 2020 study published in Molecules, Chen et al. developed a stable-isotope dimethyl labeling coupled with phosphatase dephosphorylation (DM+deP) strategy for site-specific absolute quantification of phosphorylation degree — and validated the approach directly against AQUA synthetic peptide standards with SRM mass spectrometry.
Background: Accurate determination of site-specific phosphorylation stoichiometry remains technically challenging. Most large-scale phosphoproteomics studies report only relative changes, missing the absolute occupancy information that is essential for understanding signaling network dynamics.
Approach: The team split each peptide sample into two aliquots. One aliquot was directly labeled with light formaldehyde (CH₂O). The other was first treated with phosphatase to remove all phosphate groups, then labeled with heavy formaldehyde (CD₂O). By comparing the heavy-to-light ratio of the nonphosphorylated peptide, the absolute phosphorylation degree at each site was calculated. The method was validated using synthetic peptides with defined phosphorylation degrees (20%, 40%, 60%, and 80%) and cross-validated against AQUA-based absolute quantification using synthetic phosphorylated and nonphosphorylated peptide standards under SRM mode.
Key Findings:
- The DM+deP method achieved excellent linearity (R² = 0.9971) across the tested phosphorylation degree range using synthetic peptide mixtures
- For α-S1-casein Ser130, the DM+deP approach measured phosphorylation degree at 97.2%, in close agreement with AQUA-SRM absolute quantification (98.9%), confirming assay accuracy
- The method was applied to monitor Hsp27 Ser82 phosphorylation changes in HepG2 hepatocellular carcinoma cells under oxidative stress (t-BHP treatment), demonstrating biologically relevant modulation of phosphorylation stoichiometry
- Results from DM+deP were comparable to conventional stable-isotope dimethyl labeling with TiO₂ enrichment for relative quantification, while providing the additional absolute stoichiometry information
Significance: This study demonstrates that absolute quantification of site-specific phosphorylation degree is achievable using isotope labeling strategies combined with targeted MS, and validates AQUA-based methods as a gold-standard reference for phosphorylation stoichiometry determination. The approach is directly extensible to other modification types.
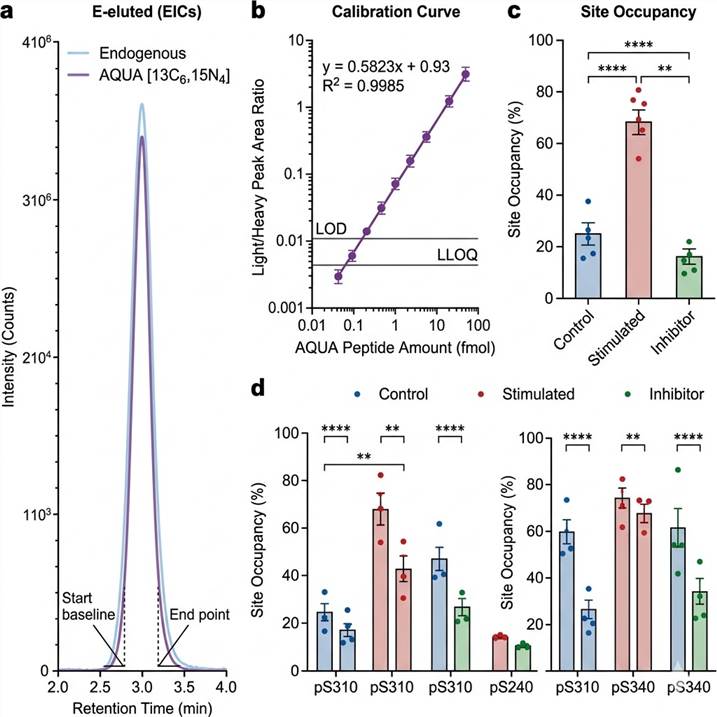
Figure from Chen et al. (2020). Validation of site-specific phosphorylation degree quantification by DM+deP approach against AQUA-SRM absolute quantification. (CC BY)
Related Services
Our absolute quantification service is part of a comprehensive PTM quantification platform. These complementary services can be used independently or integrated into a multi-phase quantitative project.
FAQs
What is the difference between absolute and relative quantification of modified peptides?
Relative quantification measures the fold-change of a modified peptide between two or more conditions (e.g., "2.5-fold increase in treatment vs. control"). Absolute quantification assigns a precise molar value (e.g., "12.3 fmol phosphorylated peptide per mg protein"), enabling direct comparison across experiments, laboratories, and biological contexts.
What is an AQUA peptide and how does it enable absolute quantification?
An AQUA (Absolute QUAntification) peptide is a synthetic peptide labeled with stable heavy isotopes (¹³C, ¹⁵N) at one or more amino acid residues. When spiked into a sample at a known concentration, it serves as an internal standard that is chemically and chromatographically identical to the endogenous target peptide but distinguishable by mass. The ratio of endogenous signal to AQUA signal gives the absolute quantity.
Which mass spectrometry platforms are used for quantification?
We use both high-resolution Orbitrap platforms for parallel reaction monitoring (PRM) and triple-quadrupole platforms for selected reaction monitoring (SRM/MRM). PRM offers superior selectivity for modified peptides in complex backgrounds; SRM provides higher multiplexing capacity for large target panels.
How many modified peptide targets can be quantified in a single experiment?
The multiplexing capacity depends on the platform. PRM methods can quantify 15–30 targets per injection, while SRM methods can accommodate 50–200+ transitions. For larger panels, multiple scheduled acquisition methods are used.
What is the typical limit of detection for modified peptide quantification?
Our AQUA-PRM and AQUA-SRM methods achieve limits of detection in the low- to mid-attomole range on column, with linear quantification spanning 3–5 orders of magnitude, depending on the specific peptide and matrix.
Can you quantify phosphorylation stoichiometry or site occupancy?
Yes — when both the modified and unmodified forms of a target peptide can be monitored, we calculate site occupancy as the ratio of modified peptide to total (modified + unmodified) peptide. This requires matched AQUA standards for both forms to correct for differential ionization efficiency.
What sample amount is needed for absolute quantification?
For most mammalian cell samples, ≥1 × 107 cells is recommended. For tissues, ≥10 mg wet weight. Required amounts depend on the abundance of the target modification and the complexity of the sample matrix. Please contact us for project-specific guidance.
How long does a typical absolute quantification project take?
A standard project (10–20 targets, 10–20 samples) typically completes in 6–8 weeks, including AQUA peptide synthesis (3–4 weeks), sample processing and LC-MS acquisition (1–2 weeks), and data analysis and reporting (1 week). Timelines vary with target number, sample count, and enrichment requirements.
References
- Chen S-H, Lin Y-C, Shih M-K, Wang L-F, Liu S-S, Hsu J-L. LC-MS Quantification of Site-Specific Phosphorylation Degree by Stable-Isotope Dimethyl Labeling Coupled with Phosphatase Dephosphorylation. Molecules. 2020;25(22):5316.
- Bezstarosti K, Van der Wal L, Doff WAS, Demmers JAA. Parallel reaction monitoring targeted mass spectrometry as a fast and sensitive alternative to antibody-based protein detection. Front Anal Sci. 2024;4:1397810.
For research use only. Not for use in diagnostic procedures.