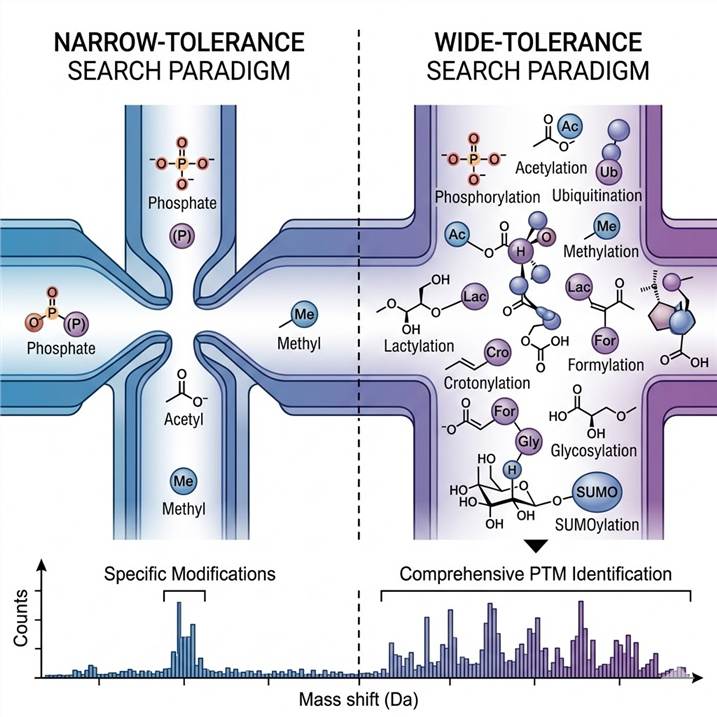
How Open-Search PTM Discovery Expands Your View of the Modificome
Traditional PTM analysis uses a "closed search" strategy: the search engine is given a fixed list of expected modifications (e.g., phosphorylation +79.97 Da, oxidation +15.99 Da) and only peptides with those exact mass shifts are considered. While efficient for targeted studies, this approach systematically excludes everything outside the predefined list — including novel modifications, unexpected adducts, and cross-species PTM variation.
Open-search (also called unrestricted or wide-tolerance searching) removes this constraint entirely. By setting the precursor mass tolerance to hundreds of Daltons, the search engine evaluates every possible mass shift between the measured precursor mass and the theoretical peptide mass. Each mass shift is a potential modification signature. The result is a comprehensive mass shift histogram — a snapshot of every modification present in your sample, both anticipated and unanticipated.
What this means for your research:
- Hypothesis-free discovery — No need to predefine which modifications matter. The data reveals what is actually present.
- Emerging PTM capture — Novel modifications such as lysine lactylation, crotonylation, 2-hydroxyisobutyrylation, and other recently characterized PTMs are detected alongside well-studied modifications in the same analysis. This is especially valuable for researchers exploring the growing landscape of emerging and rare PTMs, including those cataloged in our Rare PTM Library.
- Cross-species and cross-condition flexibility — Organisms with divergent PTM biology (e.g., bacteria, plants, non-model species) often carry modifications not represented in standard human-centric modification databases. Open search adapts without requiring custom database construction.
- Artifact detection and data integrity — Unexpected chemical adducts from sample processing, buffer components, or drug metabolites are captured and can be distinguished from biological modifications through appropriate controls, strengthening data reliability.
Rigorous False Discovery Control for Confident Identification
A common concern with open-search strategies is the risk of inflated false discovery rates. With thousands of possible mass shifts evaluated simultaneously, the search space is orders of magnitude larger than conventional closed searches. Our pipeline addresses this challenge through a multi-layered quality control framework.
Multi-Stage FDR Management
Stage 1 — Target-decoy search at the peptide level: Every open search is performed against a concatenated target-decoy database. False discovery rates are estimated using the standard target-decoy approach, with stringent FDR thresholds (≤1% at the peptide level, ≤1% at the protein level).
Stage 2 — Mass shift aggregation and PTM-Shepherd profiling: Identified mass shifts are aggregated and clustered using PTM-Shepherd or equivalent tools. Mass shifts that cluster at low abundance or fall outside expected chemical mass ranges are flagged for additional scrutiny.
Stage 3 — Closed-search cross-validation: Mass shifts corresponding to known PTMs are cross-validated in a secondary narrow-window search. Novel mass shifts proceed to orthogonal validation (PRM-targeted MS/MS or synthetic peptide matching) before being reported as confident identifications.
Site Localization Confidence
For every modified peptide, we compute site localization probabilities using validated algorithms (ptmRS, PhosphoRS, or equivalent). Sites with localization probability less than 0.75 are reported as "ambiguous" with clear annotation. This transparency ensures you can confidently assign modification sites for downstream biological interpretation.
Contamination and Artifact Filtering
Common artifacts (keratin oxidation, plasticizer adducts, buffer-derived modifications) are identified through blank control matching and excluded from final results. A contamination report is included in every deliverable package. For deeper characterization of known modifications, our Global PTM Profiling Service provides complementary targeted coverage of established modification classes.
Compatible Sample Types and Project Scope
Our open-search PTM discovery pipeline accepts a wide range of biological sample types, with minimal input requirements optimized for even limited or precious samples.
Accepted Sample Types
| Sample Type |
Minimum Requirement |
Notes |
| Cultured cells (mammalian) |
≥1 × 107 cells |
Adherent or suspension; optional metabolic labeling available |
| Cultured cells (yeast, bacteria) |
≥5 × 107 cells |
Higher input compensates for lower protein per cell |
| Tissues |
≥10 mg wet weight |
Snap-frozen, RNAlater-stabilized, or FFPE |
| Biofluids (plasma/serum) |
≥100 μL |
High-abundance protein depletion optional |
| Biofluids (CSF, urine, supernatant) |
≥500 μL |
May require concentration step |
| Subcellular fractions |
≥50 μg protein equivalent |
Nuclear, cytoplasmic, membrane, mitochondrial |
| Extracellular vesicles |
≥1 × 1010 particles |
Ultracentrifugation or kit-purified |
| Immunoprecipitated protein complexes |
≥5 μg protein per pull-down |
On-bead digestion supported |
Optional Enrichment Modules
For projects targeting low-abundance modifications or specific PTM classes, our open-search pipeline can be preceded by affinity-based enrichment:
- Phosphopeptide enrichment (TiO₂, IMAC)
- Acetylated peptide enrichment (pan-acetyl antibody)
- Ubiquitinated peptide enrichment (diGly remnant antibody)
- Glycopeptide enrichment (lectin affinity, HILIC)
- PTM-specific antibody arrays
These enrichment steps narrow the biological focus without compromising the open-search philosophy — the search remains unrestricted, simply starting from an enriched peptide population.
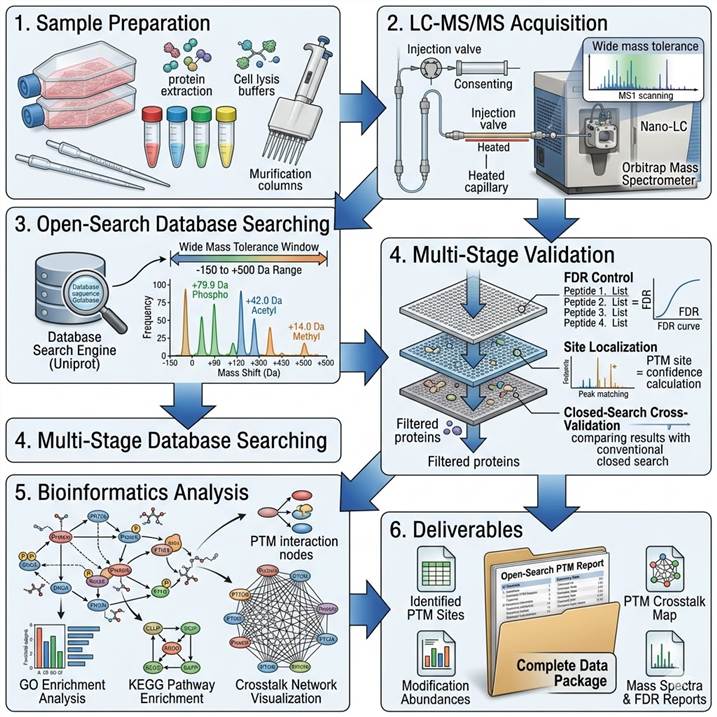
End-to-End Workflow: From Sample to Pathway-Ready Data
Step 1: Sample Preparation and QC
Samples are processed under standardized conditions. Protein extraction, reduction, alkylation, and digestion (trypsin/Lys-C or alternative proteases) follow established protocols. Each sample batch includes a process blank and a pooled QC sample. Protein integrity (SDS-PAGE), concentration (BCA assay), and digestion efficiency are documented.
Step 2: LC-MS/MS Acquisition
Peptides are separated by nano-flow reverse-phase chromatography (C18, 75 μm × 25 cm column, 120-min gradient) coupled to a high-resolution Orbitrap or Q-TOF mass spectrometer. Data-dependent acquisition is performed with high-resolution MS1 and MS2 scans, with precursor tolerance for open search set to −150 to +500 Da.
Step 3: Open-Search Database Searching
Raw files are processed through our open-search pipeline: primary open search (wide precursor tolerance, narrow fragment tolerance), mass shift clustering via PTM-Shepherd, and assignment of mass shifts to known (UniMod) or candidate novel modifications.
Step 4: Multi-Stage Validation
Target-decoy FDR filtering (≤1% PSM, ≤1% protein), site localization scoring, closed-search reconfirmation for known PTM mass shifts, and blank subtraction for artifact identification ensure data quality at every level.
Step 5: Bioinformatics Analysis
PTM landscape summary (mass shift histogram, modification class distribution), differential abundance analysis, Gene Ontology and KEGG pathway enrichment, PTM crosstalk analysis, and interactive data visualization. For deeper functional interpretation, our PTM Bioinformatics Analysis service provides dedicated pathway mapping and crosstalk prediction.
Step 6: Deliverables
Raw MS data files, search results (peptide-spectrum matches, protein groups, modification table), mass shift histogram and PTM landscape summary, site localization confidence scores, differential expression tables, bioinformatics report, contamination and QC report, and a scientist consultation session.
Representative Results: What Open-Search PTM Discovery Delivers
The output of an open-search PTM discovery experiment extends far beyond a simple list of modified peptides. Our data packages provide a comprehensive view of the modificome, enabling researchers to assess the breadth of modifications present, prioritize targets for follow-up, and directly transition into biological interpretation.
Representative data outputs from our open-search PTM discovery pipeline. Left: Mass shift histogram with annotated PTM classes. Center: PTM class distribution pie chart. Right: Differential modification abundance heatmap across conditions.
Key data components included in every deliverable package:
- Mass shift histogram — Every detected mass shift plotted and annotated against known PTM masses (UniMod). Unidentified mass shifts are flagged for orthogonal validation.
- PTM landscape summary — Classification of all identified modifications by type (phosphorylation, acetylation, ubiquitination, methylation, formylation, etc.) with relative abundance distribution.
- Site localization confidence — Per-site localization probabilities enable confident assignment of modification positions within each peptide sequence.
- Differential modification analysis — For multi-condition experiments, quantitative comparisons reveal condition-specific PTM regulation patterns.
- Pathway enrichment maps — Modified proteins mapped to biological pathways, highlighting functional networks affected by PTM changes.
Why Choose Our Open-Search PTM Discovery Service
Unbiased, Not Prescribed
Unlike standard PTM profiling that restricts the search space to a predefined list, our workflow evaluates every measurable mass shift. This approach routinely uncovers modifications that closed searches miss — from unexpected lysine acylations to novel redox modifications.
True Emerging PTM Discovery
The field of PTM biology is expanding rapidly. Modifications such as lysine lactylation, beta-hydroxybutyrylation, crotonylation, and 2-hydroxyisobutyrylation have only gained recognition in recent years. Our open-search pipeline identifies these emerging modifications alongside established ones — and continues to detect the next generation of novel PTMs as the field evolves.
Integrated Bioinformatics, Not Just a Spectrum List
We deliver pathway-ready biological interpretation, not raw search outputs. Every project includes PTM-functional enrichment, crosstalk analysis where applicable, and a consultative review session with our scientists. For comprehensive multi-modality PTM surveys, our Pan PTM Proteomics platform extends this capability across multiple modification classes simultaneously.
Deep PTM Expertise Across Modification Classes
From the most abundant phosphorylation events to low-stoichiometry ubiquitination and rare modifications — our team has hands-on experience with the full breadth of known and emerging modification types. This expertise translates directly into better data quality and more meaningful biological insights.
Demonstrated Publication Track Record
Our scientists have contributed to peer-reviewed studies employing open-search PTM strategies across diverse biological contexts — from single-cell signaling analysis to disease mechanism research and drug target discovery. This publication experience ensures that every project benefits from methods that have been validated through the rigors of academic peer review.
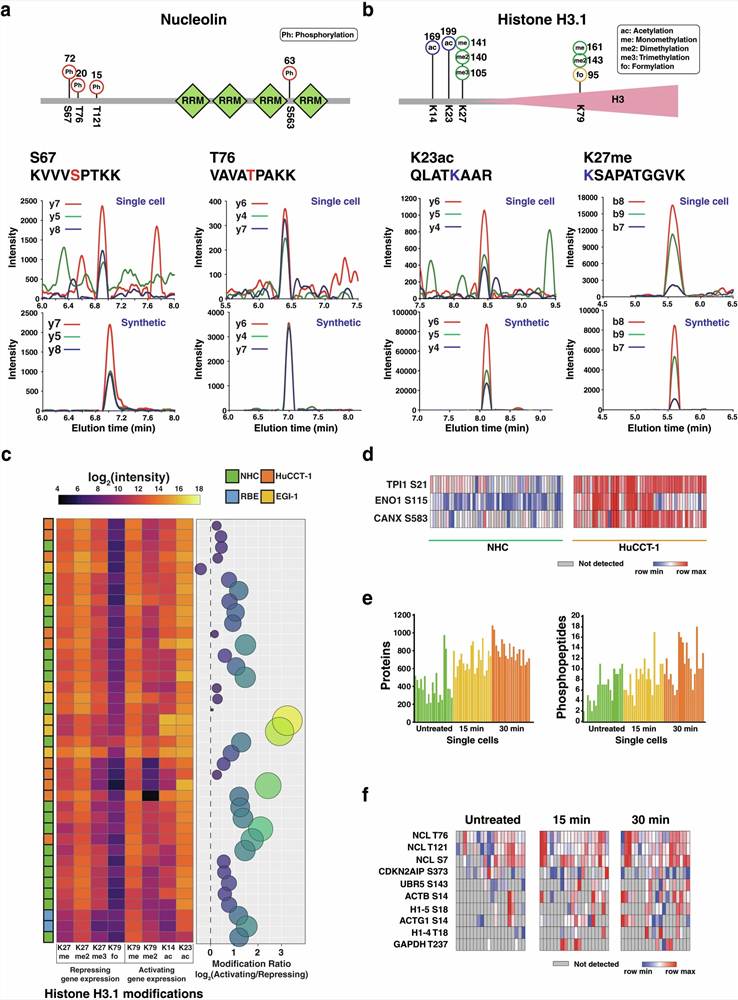
Case Study: Open Search Revealing PTM Diversity in Single Cells
In a 2024 study published in Communications Biology, Mun et al. used an optimized open-search strategy within the FragPipe platform to profile post-translational modifications at single-cell and single-organelle resolution — an analytical challenge that demands maximum sensitivity and unbiased modification coverage.
Background: Single-cell proteomics has traditionally focused on protein identification and quantification, with very limited PTM characterization. The authors aimed to determine whether open-search strategies could detect PTM diversity at the single-cell level — a scenario where sample amounts are minimal and false discovery control is especially critical.
Approach: The team processed single cells and single nuclei using an optimized DIA/diaPASEF workflow on a TIMS-TOF platform. Data were analyzed using FragPipe's open-search mode with wide precursor mass tolerance, enabling unrestricted detection of all measurable mass shifts. PTM-Shepherd was used for mass shift clustering and interpretation.
Key Findings:
- 192 phosphorylated peptides were identified from single-cell samples, including known signaling phosphosites on kinases and transcription factors
- 16 lysine-formylated peptides were detected — a relatively less common modification that would likely be excluded from standard closed-search parameter sets
- Methylation variants (mono-, di-, and trimethylation) were distinguished on lysine and arginine residues
- Single amino acid polymorphism variant peptides including the clinically relevant KRAS G12D mutation were detected concurrently with PTMs
- Single-nucleus analysis revealed epigenetic drug-induced changes in nuclear PTM profiles
Significance: This study demonstrates that open-search PTM discovery can simultaneously capture multiple modification classes, variant peptides, and unexpected modifications — providing a comprehensive view of the cellular modificome that targeted approaches cannot match.
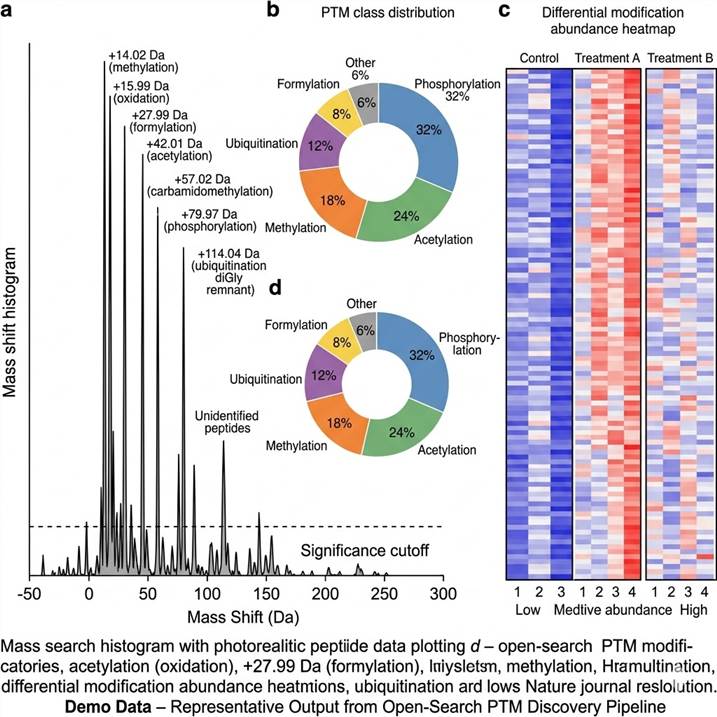
Figure 4 from Mun et al. Identification of post-translational modifications in single cells using open-search strategy. (CC BY 4.0)
Related PTM Discovery Services
Our open-search service is one component of a comprehensive PTM discovery and analysis platform. These complementary services can be used independently or integrated into a multi-phase project for deeper biological insight.
FAQs
How does open-search PTM discovery differ from standard PTM analysis?
Standard PTM analysis restricts the search to a predefined list of modifications (e.g., phosphorylation, acetylation, oxidation). Open search evaluates all possible mass shifts, detecting modifications both known and unknown without requiring prior specification. This makes it ideal for exploratory studies, novel modification discovery, and samples from non-model organisms.
What is the false discovery rate for open-search results?
We apply a multi-stage FDR control framework: target-decoy searching at ≤1% FDR at the peptide and protein level, mass shift clustering to filter sporadic matches, and closed-search cross-validation for known modifications. Novel modifications receive additional orthogonal validation before reporting.
Can open search detect very low-abundance modifications?
Yes — the sensitivity of open search depends on MS acquisition quality rather than search parameters. Our high-resolution Orbitrap and TIMS-TOF platforms provide the deep coverage needed to detect low-stoichiometry modifications. For very low-abundance targets, optional enrichment modules (phosphopeptide enrichment, antibody-based enrichment) can be added upstream.
What types of novel modifications has your service identified?
Our open-search pipeline has successfully identified a wide range of modifications beyond the conventional set, including lysine formylation, 2-hydroxyisobutyrylation, beta-hydroxybutyrylation, crotonylation, glutarylation, and various redox-derived cysteine modifications. We also detect unexpected adducts from drug metabolites and environmental exposures.
How much sample material is needed?
For standard mammalian cell samples, ≥1 × 107 cells is recommended. Tissues: ≥10 mg. Biofluids: ≥100 μL plasma/serum. Minimum input requirements vary by sample type and project scope — please consult with our team for project-specific guidance.
What bioinformatics support is included?
Every project includes PTM landscape profiling, differential abundance analysis (for multi-condition studies), Gene Ontology and KEGG pathway enrichment, PTM site motif analysis, and a one-on-one data walk-through with our scientists. Optional add-on modules include PTM crosstalk prediction and structural modeling.
Do you offer open search for non-human samples?
Absolutely. Open search is especially powerful for non-model organisms, where modification databases are often incomplete. Our pipeline uses the organism's own protein database, and the unrestricted search strategy ensures modifications specific to that organism are captured regardless of database representation.
How long does a typical open-search PTM discovery project take?
A standard project (10–20 samples, single condition) typically completes in 4–6 weeks from sample receipt to final deliverable. Timelines vary with project complexity, sample number, and optional enrichment modules.
References
- Mun D-G, Bhat FA, Joshi N, et al. Diversity of post-translational modifications and cell signaling revealed by single cell and single organelle mass spectrometry. Commun Biol. 2024;7:884.
- Freestone J, Noble WS, Keich U. Analysis of Tandem Mass Spectrometry Data with CONGA: Combining Open and Narrow Searches with Group-Wise Analysis. J Proteome Res. 2024;23(6):2127-2138.
- Moltó E, Pintado C, Andrade Louzada R, et al. Unbiased Phosphoproteome Mining Reveals New Functional Sites of Metabolite-Derived PTMs Involved in MASLD Development. Int J Mol Sci. 2023;24(22):16172.
For research use only. Not for use in diagnostic procedures.