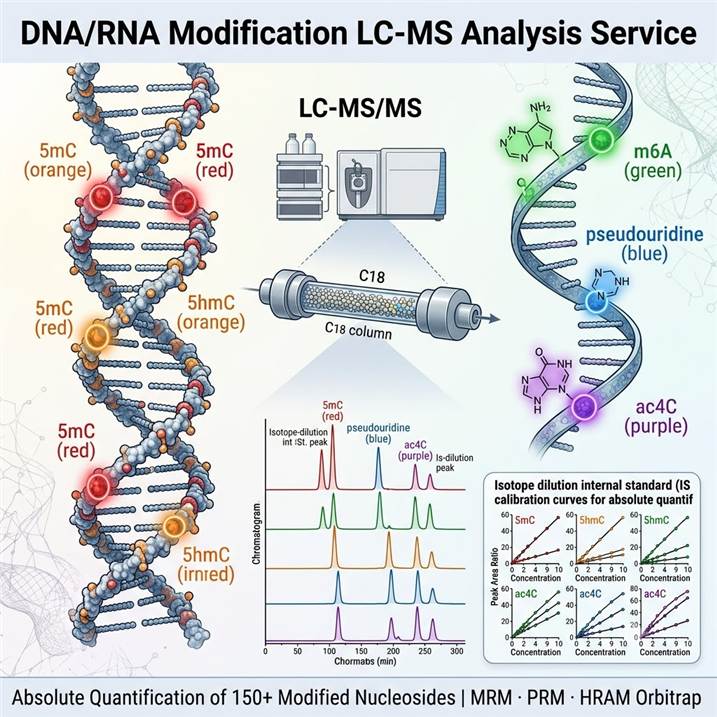
Meeting the Challenge of DNA and RNA Modification Analysis by LC-MS
Chemical modifications of DNA and RNA constitute a fundamental regulatory layer that extends the information capacity of nucleic acids beyond the primary sequence. In DNA, 5-methylcytosine (5mC) and its oxidized derivatives (5hmC, 5fC, 5caC) regulate gene expression through epigenetic silencing and active demethylation, while N6-methyladenine (6mA) has emerged as a conserved DNA modification in eukaryotes. In RNA, over 170 post-transcriptional modifications have been identified — with m6A, pseudouridine (Ψ), m5C, and m1A representing the most abundant internal modifications — that collectively regulate RNA splicing, stability, translation, and nuclear export through a dynamic network of writer, reader, and eraser proteins.
Why LC-MS is the Method of Choice for Nucleic Acid Modification Analysis
Liquid chromatography-mass spectrometry (LC-MS) offers unique advantages for nucleic acid modification analysis that complement sequencing-based approaches. LC-MS provides direct, absolute quantification of modified nucleosides without the amplification bias, antibody cross-reactivity, or reverse transcription artifacts that can affect sequencing methods. The technique simultaneously detects and quantifies multiple modifications in a single analytical run, delivers precise stoichiometric measurements (modification percentage relative to unmodified nucleosides), and is equally applicable to both DNA and RNA modifications using analogous analytical workflows. Our platform deploys high-resolution accurate mass (HRAM) Orbitrap systems for broad-spectrum modification discovery and triple quadrupole (QQQ) systems operating in MRM/PRM mode for targeted absolute quantification with isotope-labeled internal standards, providing the analytical flexibility to match the specific requirements of each project. For detailed methodology on DNA base modification analysis, our DNA Base Modification Quantification LC-MS service provides specialized workflows for epigenetic DNA modification analysis.
Our DNA/RNA modification LC-MS platform integrates multiple analytical strategies — from broad-spectrum modified nucleoside screening in epitranscriptomic discovery to targeted MRM quantification of specific modifications in clinical samples — allowing researchers to select the analytical depth that matches their biological question. Whether profiling the m6A epitranscriptome of stem cell differentiation, quantifying 5hmC levels in tumor tissues, measuring tRNA modification dynamics in metabolic disease, or assessing oxidative DNA damage in aging studies, our services deliver the precise, absolute quantification data needed for confident biological interpretation.
Our DNA/RNA Modification LC-MS Analysis Service Portfolio
We offer a structured portfolio of DNA and RNA modification analysis services designed to address specific research objectives — from broad-spectrum modified nucleoside screening to targeted absolute quantification of individual modifications. The table below maps common research goals to our recommended service modules.
| Research Objective |
Recommended Service |
Key Technology |
| Comprehensive RNA modification profiling (epitranscriptome discovery) |
RNA Modification LC-MS/MS Discovery Profiling |
HRAM Orbitrap full-scan, nucleoside-to-base ion ratio confirmation, 50+ modification library |
| Absolute quantification of specific RNA modifications by MRM/PRM |
Targeted RNA Modification Quantification |
QQQ MRM with isotope-labeled internal standards (13C, 15N), LOD down to fmol level |
| DNA methylation and hydroxymethylation analysis (5mC, 5hmC, 5fC, 5caC) |
DNA Base Modification Quantification |
LC-MS/MS with enzymatic hydrolysis, isotope dilution, % modification calculation |
| m6A epitranscriptome profiling and stoichiometry analysis |
m6A Modification LC-MS Analysis |
m6A-specific LC-MS/MS, m6A/A ratio quantification, parallel sequencing integration |
| tRNA modification analysis (codon usage, decoding, stability) |
tRNA Modification LC-MS Analysis |
tRNA isolation, RNase digestion, LC-MS/MS for modified nucleosides |
| Oxidative DNA/RNA damage assessment (8-oxodG, 8-oxoG, etheno adducts) |
DNA/RNA Adductomics and Oxidative Damage Assay |
LC-MS/MS with cleanup/enrichment, trace-level adduct quantification |
Each service module is available independently or can be combined into an integrated nucleic acid modification characterization workflow. For studies requiring high-throughput multiplexed modification detection across large sample sets, our DNA/RNA Modification Immunoassays platform provides complementary antibody-based screening capabilities alongside LC-MS validation.
DNA and RNA Modification Detection Capabilities
The following table summarizes key DNA and RNA modifications detectable by our LC-MS platform, with representative detection parameters for each modification class. Detection limits are matrix-dependent and validated for each sample type.
| Modification |
Nucleic Acid |
Mass Shift (Da) |
Typical Detection Mode |
Representative LOD |
| 5-Methylcytosine (5mC) |
DNA |
+14.016 |
MRM (QQQ) / HRAM (Orbitrap) |
0.01% of C |
| 5-Hydroxymethylcytosine (5hmC) |
DNA |
+30.011 |
MRM (QQQ) / HRAM (Orbitrap) |
0.001% of C |
| 5-Formylcytosine (5fC) |
DNA |
+28.006 |
HRAM (Orbitrap) / MRM |
0.0001% of C |
| 5-Carboxylcytosine (5caC) |
DNA |
+44.001 |
HRAM (Orbitrap) |
0.0001% of C |
| N6-Methyladenine (6mA) |
DNA |
+14.016 |
MRM (QQQ) / HRAM |
0.0001% of A |
| 8-Oxo-2'-deoxyguanosine (8-oxodG) |
DNA |
+15.995 |
MRM (QQQ) with enrichment |
1 fmol on-column |
| N6-Methyladenosine (m6A) |
RNA |
+14.016 |
MRM (QQQ) / HRAM |
0.01% of A |
| Pseudouridine (Ψ) |
RNA |
0 (isomer) |
HRAM (Orbitrap) / specific MRM |
0.01% of U |
| 5-Methylcytidine (m5C) |
RNA |
+14.016 |
MRM (QQQ) / HRAM |
0.01% of C |
| N1-Methyladenosine (m1A) |
RNA |
+14.016 |
MRM (QQQ) / HRAM |
0.01% of A |
| N4-Acetylcytidine (ac4C) |
RNA |
+42.011 |
MRM (QQQ) / HRAM |
0.01% of C |
| 7-Methylguanosine (m7G) |
RNA |
+14.016 |
MRM (QQQ) / HRAM |
0.01% of G |
| Inosine (I) |
RNA |
0 (A→I deamination) |
HRAM (Orbitrap) / MRM |
0.001% of A |
| 2'-O-Methylation (Nm) |
RNA |
+14.016 |
HRAM (Orbitrap) / MRM |
0.1% of N |
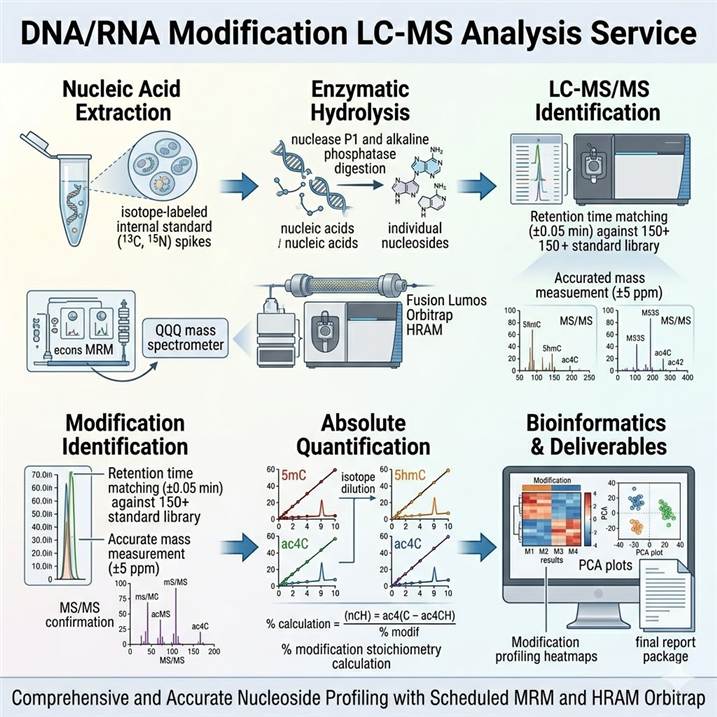
Integrated Technical Platform for DNA/RNA Modification LC-MS Analysis
Reliable nucleic acid modification analysis depends on optimized methods across the entire analytical pipeline — from nucleic acid extraction and enzymatic hydrolysis through high-resolution LC-MS acquisition to modification identification and absolute quantification. Our platform integrates best-in-class approaches at each stage, configurable to match the nucleic acid type, modification classes of interest, and required quantification precision.
Nucleic Acid Extraction and Hydrolysis
DNA and RNA are extracted using optimized protocols that preserve labile modifications while eliminating contaminants that interfere with LC-MS analysis. For RNA, we employ acidic phenol-chloroform extraction with phase separation for total RNA, followed by poly(A) selection for mRNA, tRNA isolation using size-exclusion or acid-phenol methods, or small RNA enrichment as required. DNA is extracted using proteinase K digestion and organic extraction with RNase treatment. Purified nucleic acids are enzymatically hydrolyzed to individual nucleosides using nuclease P1, phosphodiesterase, and alkaline phosphatase in a sequential digestion protocol, with digestion efficiency monitored by UV quantification and spiked internal standards. For DNA base modification analysis, our comprehensive PTM analysis platform uses validated hydrolysis and cleanup protocols optimized for each DNA modification type.
High-Resolution LC-MS/MS Acquisition
Hydrolyzed nucleosides are separated by reversed-phase liquid chromatography using C18 columns optimized for polar nucleoside retention. Mobile phases containing volatile ion-pairing agents or ammonium acetate buffers provide reproducible retention times and optimal electrospray ionization efficiency. Detection is performed on dual-platform configurations — Orbitrap HRAM systems operating at ≥60,000 resolution for full-scan discovery and accurate mass confirmation of known and unknown modifications, and triple quadrupole (QQQ) systems in MRM mode for targeted absolute quantification with isotope dilution. Scheduled MRM acquisition maximizes dwell time per transition for optimal signal-to-noise at trace levels. For m6A analysis specifically, we offer dedicated LC-MS/MS workflows optimized for m6A/A ratio quantification, as detailed on our m6A Modification LC-MS Analysis service page.
Modification Identification and Quantification
Modified nucleosides are identified by matching retention times and accurate masses (±5 ppm) against a comprehensive in-house library covering 150+ DNA and RNA modification standards, with identification confirmed by MS/MS fragmentation patterns and comparison to isotope-labeled internal standards. Absolute quantification is performed using external calibration curves with matrix-matched standards and isotope dilution with 13C- or 15N-labeled internal standards (where available), providing accurate quantification across 3–4 orders of dynamic range. Modification stoichiometry (% modification relative to the unmodified nucleoside) is calculated using the ratio of modified to unmodified nucleoside peak areas, normalized to internal standard recovery.
Quality Control and Data Validation
Each analytical batch includes system suitability standards, blank injections, calibration verification standards, and independent quality control samples at low, medium, and high modification levels. Inter-batch reproducibility is monitored through repeated analysis of pooled reference samples. For RNA modification analysis, we implement the analytical quality metrics established by the inter-laboratory standardization efforts in the field, ensuring cross-study comparability of modification abundances and stoichiometries.
DNA/RNA Modification LC-MS Analysis Workflow: From Sample to Publication-Ready Data
Step 1: Nucleic Acid Extraction and Quality Assessment
DNA or RNA is extracted using modification-preserving protocols optimized for each sample type. Nucleic acid quantity and purity are assessed by UV spectrophotometry and fluorometric methods. RNA integrity is verified by capillary electrophoresis (RIN score). Spiked internal standards (isotope-labeled nucleosides) are added at the earliest possible step to control for recovery and matrix effects throughout the workflow.
Step 2: Enzymatic Hydrolysis to Nucleosides
Purified nucleic acids are digested to individual nucleosides using a sequential enzymatic hydrolysis protocol (nuclease P1, phosphodiesterase I, alkaline phosphatase). Hydrolysis efficiency is monitored by UV absorbance at 260 nm and confirmed by the absence of oligonucleotide peaks in LC-MS chromatograms. Samples are filtered and desalted before LC-MS injection.
Step 3: LC-MS/MS Data Acquisition
Hydrolyzed nucleosides are separated by optimized C18 reversed-phase LC with gradient elution. HRAM Orbitrap full-scan acquisition (60,000–120,000 resolution) provides broad-spectrum modification discovery, while QQQ MRM acquisition delivers targeted absolute quantification with isotope dilution. Scheduled MRM windows maximize sensitivity for trace-level modifications.
Step 4: Modification Identification and Validation
Modified nucleosides are identified by retention time matching (±0.05 min) against authentic standards, accurate mass measurement (±5 ppm), and MS/MS fragmentation pattern confirmation. Unknown or unexpected modifications are characterized by accurate mass database searching and fragmentation interpretation. Each identification is validated against system suitability and QC sample criteria.
Step 5: Absolute Quantification and Stoichiometry Calculation
Absolute modification concentrations are calculated from external calibration curves with isotope dilution internal standard normalization. Modification stoichiometry (% modification = modified nucleoside / (modified + unmodified nucleoside) × 100) is calculated for each modification. Results are reported with measurement uncertainty, limits of detection, and inter-batch quality control metrics.
Step 6: Bioinformatics Analysis and Deliverables
Complete dataset including modification identification table with retention time, accurate mass, MS/MS confirmation, absolute concentration (fmol/µg nucleic acid or ng/mL), modification stoichiometry (%), inter-sample comparison statistics, and a scientist consultation session for biological interpretation of modification changes in the context of epigenetic, epitranscriptomic, or nucleic acid damage research.
DNA/RNA Modification LC-MS Analysis in Biomedical Research
Nucleic acid modification analysis by LC-MS spans a broad range of research contexts, reflecting the fundamental regulatory roles of DNA and RNA modifications in gene expression, development, and disease. Our platform is configured to support the specific requirements of each application domain, from global modified nucleoside screening in epitranscriptomic discovery to targeted quantification of specific modifications in clinical cohorts.
Cancer Epigenetics and DNA Modification Biomarkers
Global DNA hypomethylation and gene-specific hypermethylation are hallmarks of cancer genomes, with 5mC and 5hmC levels serving as diagnostic and prognostic biomarkers across multiple cancer types. Our LC-MS platform provides absolute quantification of DNA modification levels in tumor tissues, cfDNA from liquid biopsies, and urine samples, delivering the quantitative precision needed for biomarker validation and therapy monitoring. For comprehensive nucleic acid damage assessment in cancer research, our DNA/RNA Adductomics and Damage Analysis service provides parallel DNA and RNA adduct quantification from the same sample.
Epitranscriptomics and RNA Modification Dynamics
RNA modifications dynamically regulate gene expression in development, differentiation, and disease. m6A — the most abundant internal RNA modification — influences mRNA splicing, nuclear export, translation efficiency, and decay through reader protein-dependent mechanisms. Pseudouridine (Ψ) modulates RNA structure and stability, while m5C affects tRNA stability and mRNA translation. Our LC-MS platform provides the absolute quantification and stoichiometry measurements required to characterize RNA modification dynamics across biological conditions, cell types, and disease states, including dedicated workflows for mRNA and tRNA modification analysis.
Oxidative Stress and Nucleic Acid Damage
Reactive oxygen species generated during inflammation, metabolism, and environmental exposure produce a wide range of oxidized DNA and RNA lesions including 8-oxodG, 8-oxoG, etheno adducts, and lipid peroxidation products. These modifications serve as biomarkers of oxidative stress and are implicated in aging, neurodegeneration, cardiovascular disease, and carcinogenesis. Our ultra-sensitive LC-MS/MS methods with enrichment and cleanup enable quantification of trace-level oxidative damage products in tissue, blood, and urine samples for oxidative stress research.
tRNA Biology and Modification in Disease
tRNA molecules carry the highest density and diversity of RNA modifications, with over 90 distinct modifications identified across all tRNA species. These modifications regulate tRNA folding, stability, aminoacylation efficiency, and codon-anticodon interactions, with tRNA modification defects linked to mitochondrial disease, neurological disorders, and cancer. Our dedicated tRNA modification analysis workflow provides comprehensive modified nucleoside profiling from purified tRNA fractions, delivering the modification maps needed for functional tRNA biology research.
Case Study: LC-MS/MS Reveals 5-Formylcytosine Is Not a Prevalent RNA Modification in Mammalian Cells
A 2025 study by Dehnen et al. published in Nature Communications applied ultrasensitive LC-MS/MS combined with chemical-assisted sequencing to resolve a long-standing controversy about the prevalence of 5-formylcytidine (f5C) in mammalian RNA. This work demonstrates the power of LC-MS-based absolute quantification for resolving fundamental questions in epitranscriptomics and highlights the importance of orthogonal validation in RNA modification research.
Background: 5-Formylcytidine (f5C) had been reported as a prevalent RNA modification with broad regulatory functions in mammalian mRNA, based largely on pyridine-borane sequencing (f5C-seq) results. However, these sequencing-based claims lacked systematic validation by orthogonal methods, raising questions about whether f5C-seq signals genuinely reflected f5C or originated from other modifications or sequencing artifacts.
Approach: The team developed an ultrasensitive LC-MS/MS method for absolute quantification of f5C in total RNA, mRNA, and tRNA fractions from mouse embryonic stem cells (mESCs) and human cell lines. Quantification was performed using stable isotope dilution with 13C-labeled internal standards, providing absolute f5C levels as parts-per-million (ppm) relative to unmodified cytidine. Parallel analyses quantified related modifications including hm5C (5-hydroxymethylcytidine) and m5C (5-methylcytidine) for comparative assessment. The authors developed a new sequencing method (FIBo-seq) with improved specificity for f5C to cross-validate LC-MS findings.
Key Findings:
- LC-MS/MS quantification showed that f5C levels in total RNA from mESCs were only ~2–7 ppm relative to C — over 100-fold lower than previous estimates from sequencing alone
- f5C became undetectable by LC-MS/MS in Alkbh1 knockout cells, establishing that all detectable f5C in mammals is ALKBH1-dependent and restricted to mitochondrial tRNAMet
- No f5C was detected in mRNA by LC-MS/MS at the achievable limit of detection, conclusively demonstrating that f5C is not a prevalent mRNA modification in mammals
- In contrast, hm5C was detected at 7–16 ppm and m5C at 500–1,000 ppm in total RNA, providing quantitative reference ranges for these modifications
- The study demonstrated that previous f5C-seq signals in mRNA originated from misidentification of N4-acetylcytidine (ac4C) and hyper-reactive cytidines rather than genuine f5C
Significance: This study establishes rigorous LC-MS/MS quantification as an essential orthogonal validation method for RNA modification discovery, demonstrating that sequencing-based methods alone can produce false-positive assignments without absolute quantification by mass spectrometry. The finding that f5C is not a prevalent mRNA modification fundamentally revises our understanding of the mammalian epitranscriptome and highlights the critical role of LC-MS in setting quantitative standards for RNA modification research. For researchers investigating nucleic acid modifications in any biological context, our LC-MS platform provides the absolute quantification capabilities needed to validate and extend findings from sequencing-based approaches.
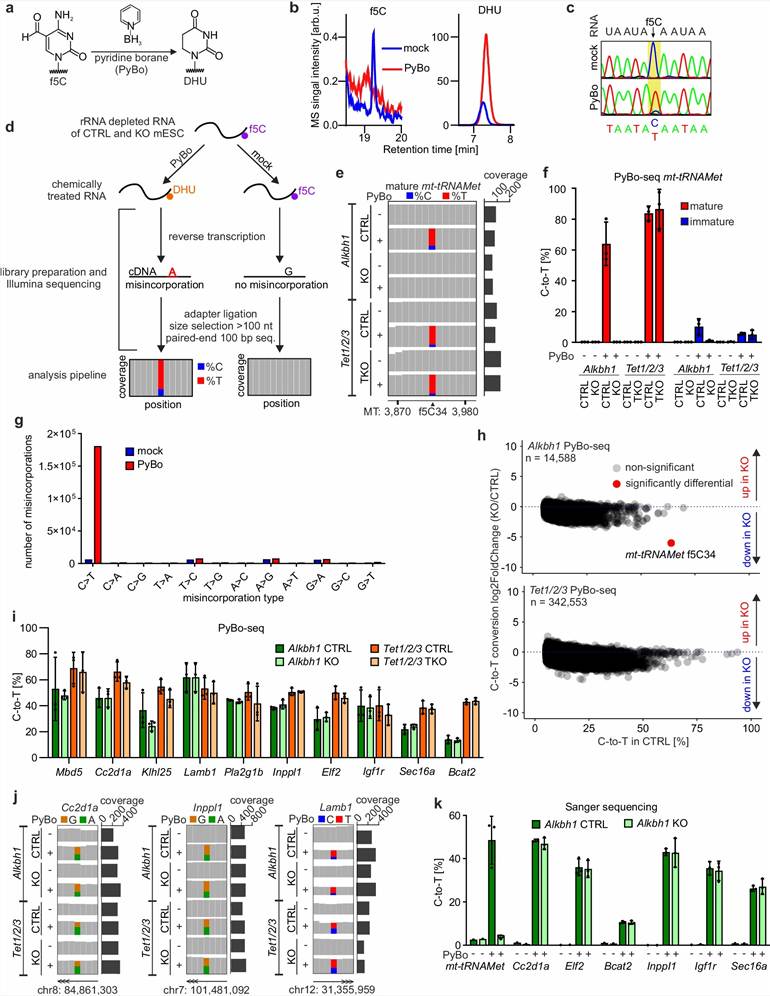
Figure 1 from Dehnen et al. (2025). Ultrasensitive LC-MS/MS quantification of 5-formylcytidine in mammalian RNA. (a) LC-MS/MS workflow for f5C quantification with isotope dilution internal standards. (b) f5C levels in total RNA and mRNA from mESCs (ppm relative to C). (c) f5C levels in Alkbh1 WT vs KO cells. (d) Quantitative comparison of f5C, hm5C, and m5C levels in total RNA. (e) FIBo-seq validation demonstrating f5C restriction to mt-tRNAMet. (f) Model showing ac4C misidentification as f5C in previous sequencing studies. (CC BY 4.0)
Representative DNA/RNA Modification LC-MS Data Outputs
Our DNA/RNA modification LC-MS analysis pipeline delivers multi-dimensional data outputs that provide a complete picture of modified nucleoside identity, absolute abundance, and stoichiometry across experimental conditions. Below are representative examples of the key data types included in every project deliverable.
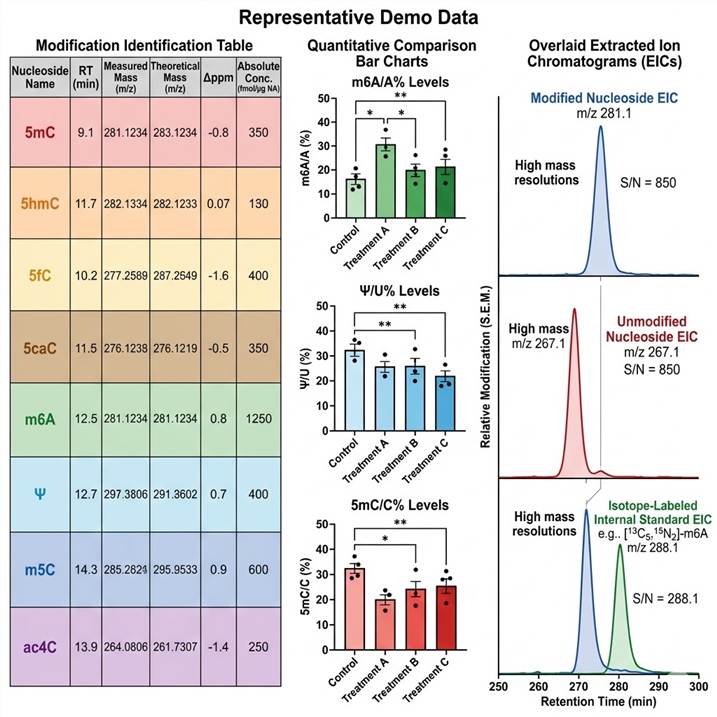
Representative DNA/RNA modification LC-MS data. (Left) Modification identification table with nucleoside name, retention time, accurate mass (Δppm), modification class, absolute concentration (fmol/µg nucleic acid), and modification stoichiometry (%). (Center) Quantitative comparison of modification levels across control and treatment conditions — significant changes highlighted by modification type. (Right) Overlaid extracted ion chromatograms (EICs) for modified nucleoside (blue), unmodified nucleoside (red), and isotope-labeled internal standard (green) showing baseline-resolved peaks with signal-to-noise ratios.
Every data deliverable includes raw chromatograms, calibration curves, QC performance metrics, and a scientist consultation session for biological interpretation of modification changes in the context of epigenetic, epitranscriptomic, or nucleic acid damage research. Custom reporting formats and data visualization options are available to match publication requirements or internal reporting standards.
Why Choose Our DNA/RNA Modification LC-MS Analysis Services
Isotope Dilution Absolute Quantification
Our LC-MS platform employs stable isotope-labeled internal standards (13C, 15N) for the majority of quantified modifications, providing the highest accuracy for absolute modification concentration and stoichiometry measurements. Isotope dilution corrects for matrix effects, extraction efficiency, and ion suppression across diverse sample types, ensuring reliable cross-study comparability.
Dual-Platform Analytical Flexibility
We deploy both Orbitrap HRAM systems for broad-spectrum modification discovery and triple quadrupole (QQQ) platforms in MRM/PRM mode for targeted high-sensitivity quantification. This dual-platform configuration provides the analytical flexibility to match the specific requirements of each project — from unbiased modified nucleoside screening to trace-level targeted quantification.
Comprehensive Modification Coverage
Our in-house library spans 150+ DNA and RNA modification standards covering all major modification classes — DNA methylation and oxidation products, RNA internal modifications (m6A, Ψ, m5C, m1A, ac4C, m7G, inosine), 2'-O-methylations, tRNA base modifications, and oxidative damage adducts. This breadth enables simultaneous multi-modality profiling from a single LC-MS run.
Cross-Platform Integration
Our nucleic acid modification analysis platform integrates LC-MS quantification with complementary approaches including antibody-based modification enrichment and immunoassays for multi-platform validation, and our bioinformatics pipeline provides integrated analysis across modification types, sample groups, and cross-study normalization for large cohort projects.
Related Services
Our DNA/RNA modification LC-MS analysis services are supported by a broader PTM and nucleic acid characterization platform offering complementary analytical capabilities across modification types and research applications.
- GlycoRNA Sequencing — Specialized analysis of glycosylated RNA molecules combining RNA biochemistry with glycan detection for emerging GlycoRNA research
- Oxidative DNA/RNA Damage Assay — Targeted quantification of oxidative damage products in DNA and RNA including 8-oxodG, 8-oxoG, and etheno adducts
- Global PTM Profiling — Broad multi-PTM discovery analysis across diverse protein modification classes for integrated multi-omics studies
- MS-Based PTM Analysis — Comprehensive mass spectrometry platform for protein-level PTM discovery, quantification, and characterization
- Modified Peptide Enrichment Services — Specialized enrichment strategies for low-abundance modified peptides and nucleic acid modifications
- PTM Bioinformatics Analysis — Advanced bioinformatics for PTM data integration, functional annotation, and nucleic acid modification pathway analysis
- PTM Proteoform Mapping — Detailed characterization of combinatorial modification patterns on individual proteoforms
- Open-Search PTM Discovery — Unbiased open-search approach for detecting unexpected modifications across nucleic acids and proteins
- Bottom-Up MS-Based PTM Analysis — Deep PTM discovery and quantitative profiling using the shotgun proteomics approach
- PTM in Biological Research — Application-focused PTM analysis solutions for specific biological research contexts
Frequently Asked Questions
What is the difference between LC-MS and sequencing-based methods for nucleic acid modification analysis?
LC-MS provides direct, absolute quantification of modified nucleosides without amplification bias or reverse transcription artifacts that can affect sequencing methods. LC-MS simultaneously detects and quantifies multiple modifications in a single run, delivers precise stoichiometric measurements (e.g., % m6A/A), and is equally applicable to both DNA and RNA. Sequencing methods provide base-resolution mapping information that LC-MS cannot directly provide. The two approaches are complementary — LC-MS provides accurate quantification and orthogonal validation, while sequencing provides positional context within individual transcripts.
What types of nucleic acid modifications can you detect and quantify?
Our in-house modification library covers over 150 DNA and RNA modifications, including DNA methylation (5mC, 5hmC, 5fC, 5caC, 6mA), RNA internal modifications (m6A, Ψ, m5C, m1A, ac4C, m7G, inosine, m6Am), 2'-O-methylated nucleosides (Nm), tRNA base and ribose modifications, and oxidative damage products (8-oxodG, 8-oxoG, etheno adducts). Custom method development is available for modifications not currently in our library.
What sample types and amounts are required for DNA/RNA modification LC-MS analysis?
For global RNA modification profiling, we recommend ≥1 µg of purified total RNA. For mRNA modification analysis, ≥100 ng of poly(A)-selected mRNA is typically sufficient. For DNA modification analysis, ≥500 ng of genomic DNA is recommended. Our platform is compatible with total RNA, mRNA, tRNA, small RNA, genomic DNA, and mitochondrial DNA extracted from cultured cells, tissues, biofluids, and FFPE samples. Smaller amounts may be acceptable for targeted MRM analysis of abundant modifications.
How do you ensure accurate absolute quantification of modifications?
Absolute quantification is performed using external calibration curves with authentic modification standards, combined with isotope dilution using 13C- or 15N-labeled internal standards spiked at the earliest possible step in the workflow. This dual approach corrects for matrix effects, ion suppression, and recovery variations across different sample types. Each analytical batch includes system suitability standards, calibration verification, and independent QC samples at multiple concentration levels.
Can you analyze both DNA and RNA modifications from the same sample?
Yes — our platform supports parallel DNA and RNA modification analysis from the same biological sample through separate nucleic acid extraction protocols optimized for each analyte class. DNA and RNA are separated during extraction using phase separation and RNase/DNase treatments, then independently hydrolyzed to nucleosides and analyzed by LC-MS/MS. This parallel approach enables integrated analysis of the DNA epigenome and RNA epitranscriptome from a single sample, providing a comprehensive view of nucleic acid modification regulation.
What is the limit of detection for different modification types?
Detection limits vary by modification type and sample matrix. For abundant modifications (m6A, m5C, Ψ) in total RNA, typical limits of detection are ~0.01% modification relative to the unmodified nucleoside (corresponding to ~1–10 fmol on-column). For rarer modifications (5fC, 5caC, 6mA in DNA), we achieve LODs down to 0.0001% using optimized MRM methods with enrichment. For oxidative damage products (8-oxodG), our ultra-sensitive methods with offline enrichment reach LODs of ~1 fmol on-column.
How does your bioinformatics analysis support DNA/RNA modification data interpretation?
Our bioinformatics pipeline includes automated modification identification through retention time and accurate mass matching against our 150+ standard library, absolute concentration and stoichiometry calculation with isotope dilution correction, inter-sample statistical analysis (differential modification analysis, PCA, hierarchical clustering), integration with public modification databases (RMBase, m6A-Atlas, MethBase) for biological context, and cross-study normalization for large cohort projects. Results are delivered with interactive visualization and a scientist consultation session.
References
- Dehnen JA, Gopanenko AV, Scholz C, Musheev MU, Niehrs C. 5-Formylcytosine is not a prevalent RNA modification in mammalian cells. Nat Commun. 2025;16:9925.
- Hermon SJ, Sennikova A, Becker S. Quantitative detection of pseudouridine in RNA by mass spectrometry. Sci Rep. 2024;14:27564.
- Hengesbach M, Chan CK, Bhandari T, Bruzel A, DeMott MS, Podoprygorina G, Sun G, Tabeling E, Cheung VG, Dedon PC, Helm M, Limbach PA. Toward standardized epitranscriptome analytics: an inter-laboratory comparison of mass spectrometric detection and quantification of modified ribonucleosides in human RNA. Nucleic Acids Res. 2025;53(17):gkaf895.
For research use only. Not for use in diagnostic procedures.