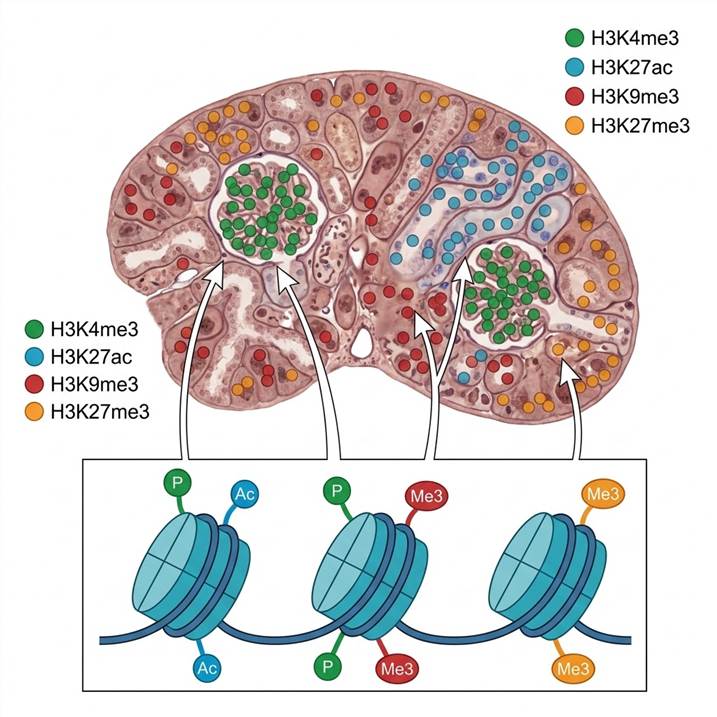
Histone Marks We Detect Spatially
Our spatial histone modification platform covers the major classes of histone PTMs that regulate chromatin structure and gene expression. Detection is achieved through three complementary modalities, each suited to different experimental needs.
Histone Methylation Marks
- H3K4me3 — active promoter mark, enriched at transcribed genes
- H3K4me1 — enhancer-associated monomethylation
- H3K9me3 — constitutive heterochromatin, silenced repeat regions
- H3K27me3 — facultative heterochromatin, Polycomb-repressed loci
- H3K36me3 — transcribed gene bodies
- H4K20me3 — pericentric heterochromatin
Histone Acetylation Marks
- H3K27ac — active enhancers and promoters
- H3K9ac — active chromatin, correlates with H3K4me3
- H4K16ac — transcriptional activation, chromatin decompaction
- Pan-acetyl-histone H3/H4 — broad acetylation profiling
Histone Phosphorylation & Other Marks
- H3S10ph — mitotic marker, immediate early gene activation
- H3S28ph — stress-responsive phosphorylation
- γH2AX (S139ph) — DNA double-strand break marker
- H3K27cr (crotonylation) — emerging active mark
All marks are detected using validated modification-specific antibodies in multiplex IF/IHC formats, or by targeted MS following histone extraction from LCM-captured tissue regions. Our histone PTM analysis service provides the full technical platform for bulk reference profiling, while the spatial workflow maps these marks onto tissue architecture. For higher-throughput screening of histone modification patterns, the histone mark antibody array service offers a complementary approach.
Applications: Epigenetic Drug Research & Tissue Heterogeneity
Epigenetic Drug Target Engagement
Epigenetic drugs — including EZH2 inhibitors (targeting H3K27me3), BET inhibitors (BRD4-acetyl histone interaction), and HDAC inhibitors (broad acetylation) — are designed to modulate histone modification states. Determining whether these drugs engage their intended targets within specific tissue compartments is critical for understanding efficacy and on-target toxicity. Our spatial histone profiling can map changes in H3K27me3 distribution across tumor and stromal compartments following EZH2 inhibitor treatment, or track H3K27ac gain at specific loci after HDAC inhibition — all within the architectural context of the tissue.
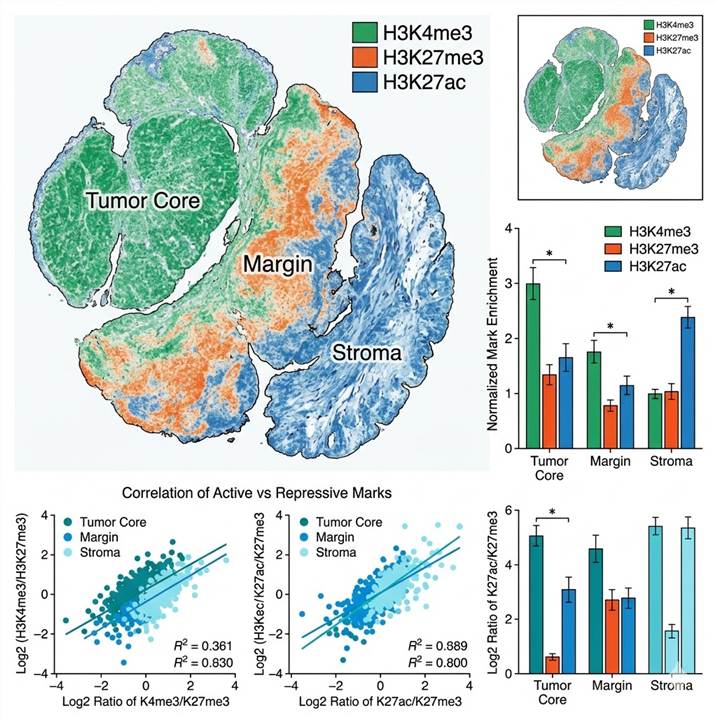
Tumor Heterogeneity and Epigenetic Subtyping
Epigenetic heterogeneity is a major driver of tumor evolution and therapy resistance. Different regions within the same tumor can harbor distinct histone modification landscapes — an H3K27me3-high, Polycomb-repressed region adjacent to an H3K4me3/H3K27ac-active, proliferative zone. Spatial histone profiling reveals these intra-tumor epigenetic compartments, enabling correlation with histopathological features, immune infiltration, and clonal architecture. This approach is supported by our epigenetic PTM research service platform.
Developmental and Tissue-Specific Epigenetics
During development and tissue homeostasis, histone modifications define cell identity. Spatial profiling can map how H3K27me3 and H3K4me3 patterns define lineage boundaries in developing organs, how H3K9me3 marks heterochromatic regions in aging tissues, or how environmental exposures alter the spatial epigenome — providing insights that bulk chromatin assays cannot resolve.
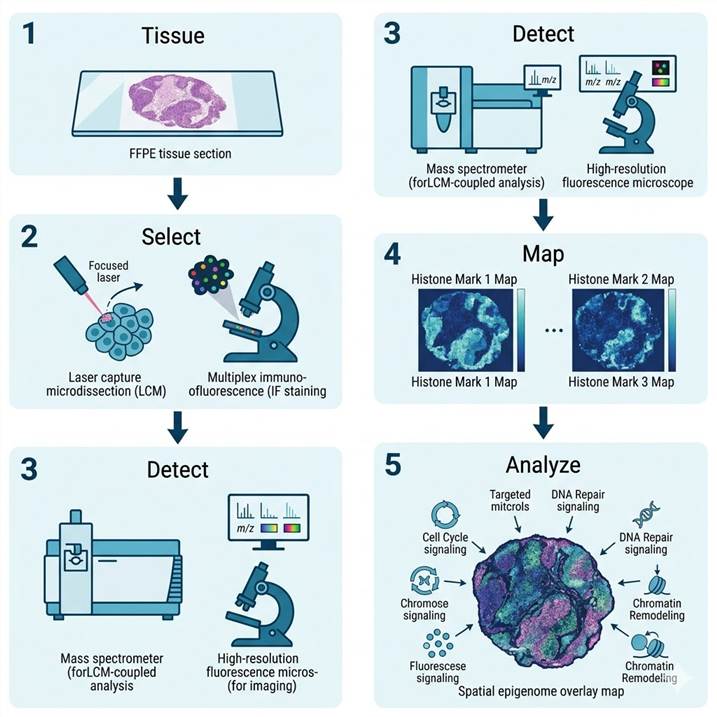
Spatial Histone PTM Profiling Workflow
The spatial histone modification workflow follows the same three-modality framework as our broader spatial PTM platform, with specific adaptations for histone extraction and detection. Typical projects run 4–8 weeks depending on modality and sample number.
Step 1: Tissue Preparation
- FFPE or frozen tissue sections (4–10 µm thick) on conductive or membrane slides
- H&E staining for pathological annotation and ROI marking
- Antigen retrieval optimization for histone modification-specific antibodies
- Histone extraction protocol selection (acid extraction for MS-based workflows)
Step 2: Modality Selection & Acquisition
- LCM-MS: laser microdissection of histone mark-dense regions
- MALDI-MSI: on-tissue histone peptide detection
- Multiplex IF: 4–8 histone modification antibodies + cell-type markers
- All data co-registered with serial H&E sections
Step 3: Histone PTM Detection
- LCM-MS: acid extraction, propionylation, tryptic digestion, LC-MS/MS
- MALDI-MSI: on-tissue digestion with histone-specific protease
- Multiplex IF: tyramide signal amplification (TSA) for signal enhancement
- Internal control marks (total H3) for normalization
Step 4: Data Processing
- LCM-MS: database search with histone variable modifications, label-free quantification
- MALDI-MSI: ion image generation for specific modified histone peptides
- Multiplex IF: spectral unmixing, cell segmentation, per-cell quantification
- Spatial correlation of multiple histone marks on same tissue section
Step 5: Interpretation
- Region-specific histone mark enrichment and differential analysis
- Co-localization analysis of active vs repressive marks
- Integration with reference bulk histone PTM data
- Publication-ready spatial heatmaps, multiplex IF overlays, and quantification
Sample Requirements for Spatial Histone Profiling
| Modality |
Recommended Sample |
Minimum Tissue |
Key Consideration |
| LCM-MS (histone extraction) |
Frozen sections (preferred) or FFPE |
5–20 mm² per region |
Acid extraction efficiency varies by tissue type |
| MALDI-MSI |
Frozen sections |
5–20 mm² per section |
On-tissue digestion optimized for histone peptides |
| Multiplex IF/IHC |
FFPE sections |
Standard slide (1–2 sections) |
Antibody validation for each histone mark required |
Note: Include matched control tissue for differential analysis. For drug treatment studies, vehicle-treated and untreated controls are required. Ship samples on dry ice with complete metadata.
Spatial Histone Profiling vs. Bulk Histone MS: Key Differences
| Dimension |
Spatial Histone Profiling |
Bulk Histone MS |
| Spatial resolution |
Tissue region to single-cell (10 µm – 1 mm) |
None (whole tissue homogenate) |
| Modification coverage |
8–20 marks per panel (targeted) |
50–200+ marks (unbiased discovery) |
| Sample input |
1–10 µg protein per region |
10–100 µg total histone extract |
| Cell-type resolution |
Yes (LCM or IF-based) |
No (bulk average) |
| Morphological correlation |
Direct (H&E overlay) |
None |
| Best suited for |
Tissue heterogeneity, drug distribution, spatial epigenomics |
Global mark discovery, stoichiometry, novel modification identification |
The two approaches are complementary. Bulk histone profiling provides comprehensive mark catalogs; spatial profiling reveals where those marks are located within the tissue. For the deepest insight, we recommend starting with bulk analysis using our quantitative histone PTM analysis and prioritizing marks for spatial follow-up.
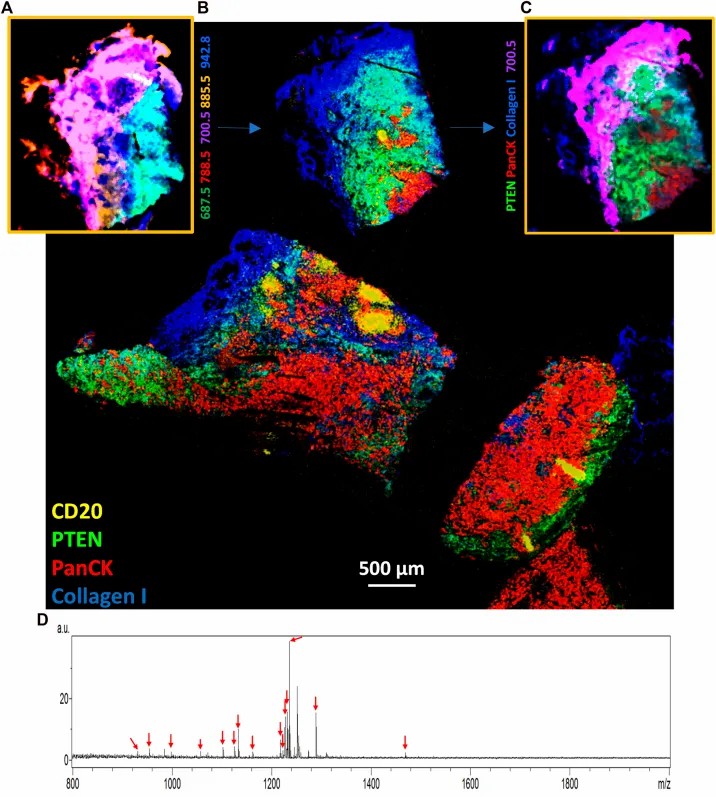
Case Study: MALDI HiPLEX-IHC for Highly Multiplexed Tissue Profiling
Background
Highly multiplexed detection of protein targets — including histone modifications — in intact tissue sections has been a long-standing goal in spatial biology. In a 2023 study published in Frontiers in Chemistry, Yagnik et al. introduced MALDI HiPLEX-IHC, a top-down spatial imaging approach that combines the multiplexing capacity of MALDI mass spectrometry with the specificity of antibody-based detection.
Methods
The approach uses photocleavable mass-tagged antibodies that are applied to tissue sections, then released by UV laser pulses and detected by MALDI-TOF MS. This allows simultaneous detection of dozens of targets from a single tissue section without the spectral overlap limitations of conventional multiplex fluorescence. The method was demonstrated on FFPE tissue sections using panels of antibodies targeting immune cell markers, signaling proteins, and chromatin-associated targets.
Results
MALDI HiPLEX-IHC achieved simultaneous detection of 12+ protein targets in single tissue sections, with the ability to co-register protein expression data with tissue morphology from serial H&E sections. The method was applied to characterize the tumor immune microenvironment, revealing spatially distinct immune cell populations and their association with chromatin state markers. Importantly, the approach is compatible with standard FFPE tissue blocks and clinically archived specimens (Yagnik et al., Front. Chem., 2023).
Conclusion
This work demonstrates that MALDI-based multiplexed antibody detection can provide deep spatial profiling of protein targets, including histone modifications, in intact tissue — a capability directly applicable to epigenetic drug development and tissue heterogeneity studies.
Source figure: Figure 3 from the original publication — MALDI HiPLEX-IHC multiplex imaging of FFPE tissue sections. (View original article)
Demo: Example Spatial Histone Modification Data
Below are representative results from spatial histone modification profiling. The workflow diagram shows the integrated pipeline from tissue section to spatial epigenome map. The multi-color spatial map illustrates differential histone mark distribution across tissue compartments.
References
- Yagnik G, Liu Z, Rothschild KJ, et al. MALDI HiPLEX-IHC: multiomic and multimodal imaging of targeted protein expression on tissue using photocleavable mass-tagged antibodies. Front. Chem. 2023; 11:1182404. DOI: 10.3389/fchem.2023.1182404
- Deng Y, Bartosovic M, Ma S, et al. Spatial-CUT&Tag: spatially resolved chromatin modification profiling at the cellular level. Science 2022; 375:681–686. DOI: 10.1126/science.abg7216
- Method of the Year 2024: spatial proteomics. Nat. Methods 2024; 21:2205. DOI: 10.1038/s41592-024-02565-3
Spatial Histone Modification Profiling: Frequently Asked Questions
What histone marks can be detected spatially?
All major histone PTM classes are compatible: methylation (H3K4me3, H3K9me3, H3K27me3, H3K36me3), acetylation (H3K27ac, H3K9ac, H4K16ac), phosphorylation (H3S10ph, γH2AX), and emerging marks such as crotonylation. The specific panel depends on the chosen modality — multiplex IF provides targeted detection of 4–8 marks, while LCM-MS can profile dozens of marks simultaneously.
Can FFPE tissue sections be used?
Yes. FFPE sections are the standard input for multiplex IF/IHC workflows and are compatible with MALDI-MSI. For LCM-MS histone extraction, frozen sections are preferred for optimal modification preservation, but FFPE can be used with appropriate antigen retrieval protocols. Our histone isolation and enrichment service provides detailed protocols for each sample type.
What spatial resolution can be achieved?
Multiplex IF provides sub-micron resolution at the single-cell level, enabling per-cell histone mark quantification. LCM-MS can isolate regions as small as 10–50 µm. MALDI-MSI achieves 10–100 µm pixel size for label-free detection. The modality choice depends on whether single-cell resolution or broad discovery coverage is prioritized.
How do you distinguish between different histone marks at the same residue?
Histone marks at the same residue (e.g., H3K27me3 vs H3K27ac) are distinguished using modification-specific antibodies that recognize the exact modification state. Each antibody is validated for specificity by dot blot and peptide competition assays. For MS-based LCM-MS, the mass shift of each modification provides unambiguous identification.
Can I correlate spatial histone data with gene expression?
Yes. LCM-captured material can be split for parallel histone PTM analysis and RNA extraction. MALDI-MSI data can be co-registered with spatial transcriptomics from serial sections. Multiplex IF panels can include both histone modification and cell-type marker antibodies for direct correlation.
How is this different from ChIP-seq or CUT&Tag?
ChIP-seq and CUT&Tag provide genome-wide maps of histone modification occupancy at specific loci, but require large cell numbers and do not preserve tissue architecture at the protein level. Our spatial histone profiling detects the actual modification state of histone proteins within intact tissue, providing complementary information about protein-level mark abundance in anatomical context. The two approaches are highly complementary.