Why Protein Monoaminylation Analysis Matters
Monoaminylation is a rapidly emerging field in PTM biology that connects neurotransmitter biochemistry with protein function regulation. Unlike classical PTMs that utilize metabolites from within the cell, monoaminylation incorporates exogenous amine substrates — serotonin, dopamine, histamine, and related molecules — into protein structure, creating a direct molecular link between the cellular microenvironment and the functional proteome.
Epigenetic Regulation Through Histone Monoaminylation
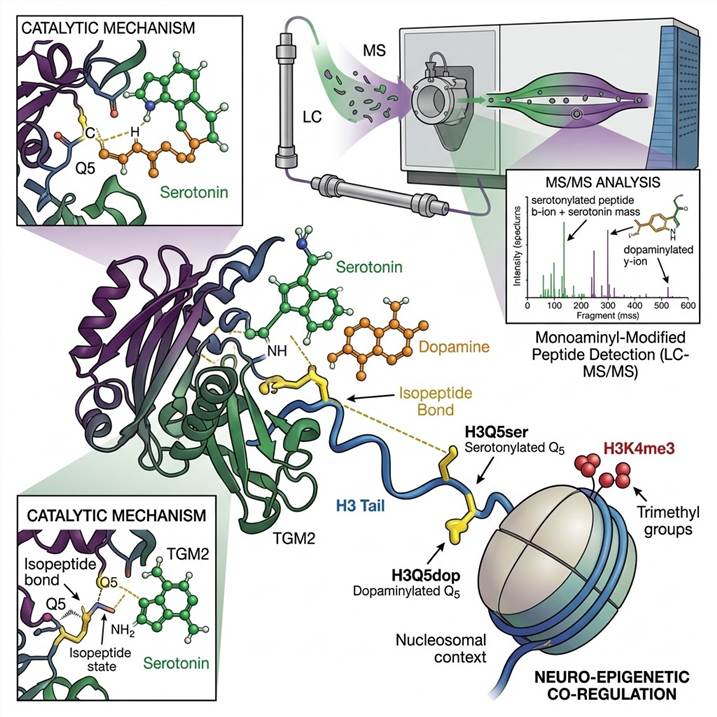
The discovery that serotonin and dopamine can be covalently attached to histone H3 at glutamine 5 (H3Q5ser and H3Q5dop) revealed an entirely new dimension of epigenetic regulation. Histone serotonylation promotes a permissive chromatin state and potentiates gene activation, while histone dopaminylation has been implicated in cocaine-induced transcriptional plasticity and addiction biology. These modifications are deposited by TGM2 and can coexist with adjacent methylation marks (H3K4me3), adding an additional layer of complexity to the histone code. The functional interplay between monoaminylation and other histone PTMs represents a frontier in understanding how neuronal activity and neurotransmitter signaling translate into stable epigenetic changes.
Non-Histone Substrates and Signaling Functions
Beyond histones, monoaminylation modifies a diverse range of proteins including small GTPases (RhoA, Rab family), metabolic enzymes (GAPDH), cytoskeletal proteins, extracellular matrix components, and signaling receptors. Serotonylation of RhoA and Rab proteins regulates platelet activation, insulin secretion, and cytoskeletal dynamics. GAPDH serotonylation couples glycolytic metabolism to CD8+ T cell effector function, revealing a direct link between the serotonergic system and anti-tumor immunity. These discoveries highlight that monoaminylation is not confined to the nucleus but operates across cellular compartments with broad functional reach.
Cancer Biology and Therapeutic Implications
TGM2 is frequently overexpressed in cancer, and monoaminylation levels are elevated in tumor tissues compared to normal adjacent tissue. Global serotonylation proteomic profiling has identified over 1,000 serotonylated proteins in cancer cells, many associated with carcinogenesis pathways. The accumulation of monoaminylated histones in cancer cell chromatin suggests that these marks contribute to the aberrant gene expression programs that drive malignancy. Targeting TGM2-mediated monoaminylation is emerging as a potential therapeutic strategy, and quantitative monoaminylation analysis is essential for understanding the scope and regulation of this modification system.
Our Approach to Monoaminylation Analysis
Protein monoaminylation presents unique analytical challenges: the modification is acid-labile, lacks pan-specific antibodies for enrichment, and occurs on glutamine residues via an isopeptide bond that requires specialized mass spectrometry detection. Our pipeline addresses each of these challenges through an integrated chemical proteomics strategy.
Chemical Probe-Based Enrichment
Because monoaminylation-specific antibodies are not widely available, we employ bioorthogonal chemical probe strategies for enrichment. Using pH-controlled chemoselective rapid azo-coupling reactions (CRACR) or alkyne-modified amine precursors, monoaminylated proteins and peptides are selectively labeled with affinity tags (biotin) and enriched via streptavidin pulldown. This antibody-free approach enables unbiased capture of the monoaminylated proteome with high specificity and sensitivity.
LC-MS/MS Detection and Site Mapping
Enriched monoaminylated peptides are analyzed on high-resolution Orbitrap platforms with optimized LC gradients for modified peptide separation. The isopeptide bond between the amine and glutamine side chain produces characteristic fragmentation signatures under HCD that enable confident identification. For comprehensive coverage, we deploy multiple proteases (trypsin, chymotrypsin, Glu-C) to maximize the sequence coverage around modification sites and improve site localization confidence.
Quantification Across Conditions
For differential monoaminylation analysis, we offer both label-free quantification (LFQ) and tandem mass tag (TMT) labeling approaches. TMT-based multiplexing enables simultaneous comparison of up to 16 samples, providing statistically robust quantification of monoaminylation changes across experimental conditions, treatment groups, or disease states. For validated targets, targeted PRM assays can be developed for precise quantification of specific monoaminylation sites.
Our Open-Search PTM Discovery service provides broader unbiased PTM profiling that complements targeted monoaminylation analysis, enabling simultaneous detection of multiple modification types in a single experiment.
Compatible Sample Types and Requirements
Our monoaminylation analysis pipeline accepts a range of sample types. The chemical probe enrichment strategy requires careful optimization for each sample matrix, and recommended starting amounts reflect the need for sufficient modified peptide material after enrichment.
| Sample Type |
Recommended Amount |
Expected Coverage |
| Cultured cells (mammalian) |
≥5 × 10⁷ cells |
Global monoaminylation profiling (hundreds of sites) |
| Tissue samples (snap-frozen) |
≥50 mg wet weight |
Tissue-specific monoaminylation site mapping |
| Histone-enriched fractions |
≥25 μg histone protein |
Site-specific histone monoaminylation (H3Q5ser, H3Q5dop) |
| Immunoprecipitated proteins |
≥10 μg per pull-down |
Targeted monoaminylation analysis of specific proteins |
| Biofluids (plasma, serum) |
≥1 mL (targeted analysis only) |
Monoaminylation of abundant plasma proteins |
Projects studying histone monoaminylation in cancer or neurobiology can be complemented by our Lactylation Analysis and Crotonylation Analysis services for integrated emerging PTM characterization.
Workflow: From Sample to Monoaminylation Data
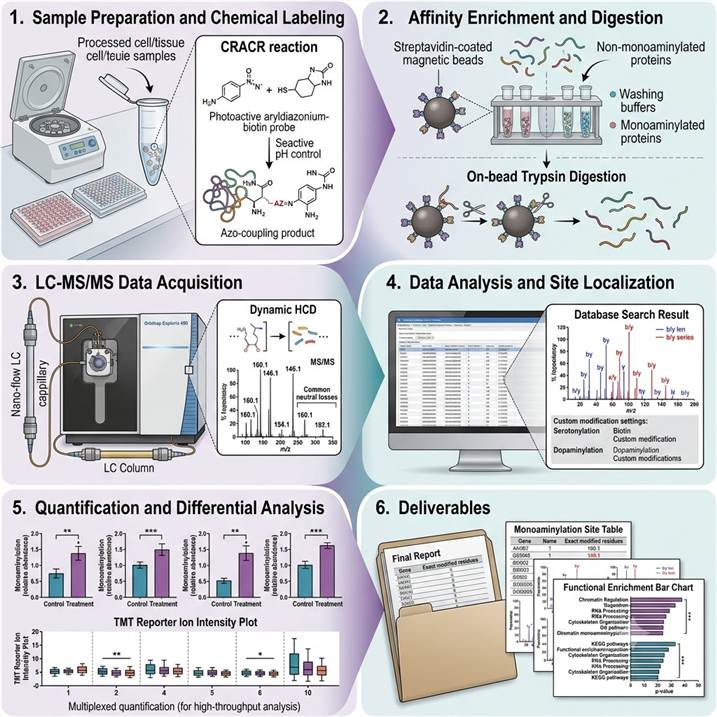
Step 1: Sample Preparation and Chemical Labeling
Proteins are extracted under conditions that preserve the labile isopeptide bond. Monoaminylated proteins are chemically labeled using bioorthogonal probes — either via CRACR chemistry (pH-controlled azo-coupling with aryldiazonium-biotin probes for direct serotonylation/dopaminylation labeling) or metabolic incorporation of alkyne-modified amine precursors. Labeling efficiency is validated by in-gel fluorescence or dot-blot before proceeding to enrichment.
Step 2: Affinity Enrichment and Digestion
Biotin-labeled monoaminylated proteins are captured on streptavidin beads under stringent washing conditions to remove non-specifically bound material. On-bead digestion with optimized proteases releases monoaminylated peptides while the biotin-probe-peptide conjugate is retained for elution. Eluted peptides are desalted and concentrated for LC-MS/MS analysis.
Step 3: LC-MS/MS Data Acquisition
Enriched monoaminyl peptides are separated using nano-flow reversed-phase chromatography with gradients optimized for modified peptide retention. Data-dependent acquisition on Orbitrap platforms with HCD fragmentation generates high-resolution MS/MS spectra. The diagnostic fragmentation signature of the monoaminyl-isopeptide bond — including the neutral loss of the amine moiety and characteristic immonium ions — is used as an additional filter for confident identification.
Step 4: Data Analysis and Site Localization
Raw MS data are searched against protein databases using custom modification definitions for serotonylation, dopaminylation, histaminylation, and related monoaminyl adducts. The isopeptide bond at glutamine residues requires specialized search parameters accounting for the amine mass shift (+C₆H₆N₂O for serotonylation, +C₈H₁₁NO₂ for dopaminylation). Site localization is validated using fragment ion coverage around the modified glutamine residue.
Step 5: Quantification and Differential Analysis
For label-free quantification, extracted ion chromatograms for monoaminylated peptides are integrated across all samples and normalized to total protein abundance. For TMT-based experiments, reporter ion intensities are extracted and normalized. Statistical analysis identifies monoaminylation sites with significant abundance changes between conditions, with appropriate multiple testing correction.
Step 6: Deliverables and Review
Monoaminylation site table with peptide sequences, modification sites, and confidence scores, annotated MS/MS spectra for each identified modification, quantitative comparison across conditions, enrichment and labeling efficiency QC report, and a scientist consultation session for biological interpretation and experimental follow-up planning.
For comprehensive emerging PTM discovery beyond monoaminylation, our Pan-PTM Proteomics service provides integrated multi-PTM profiling across diverse modification classes.
Why Choose Our Protein Monoaminylation Analysis Service
Specialized Chemical Proteomics Expertise
Monoaminylation analysis requires specialized knowledge of bioorthogonal chemistry, acid-labile modification handling, and custom search parameter configuration that extends beyond standard PTM proteomics workflows. Our team has deep expertise in chemical probe design, affinity enrichment optimization, and monoaminyl adduct mass spectrometry characterization.
Comprehensive Modification Coverage
We offer detection and site mapping for all major monoaminyl modifications: serotonylation (5-HT), dopaminylation (dopamine), histaminylation (histamine), and related biogenic amine modifications. Our chemical probe toolkit is continuously updated to incorporate the latest advances in monoaminylation enrichment technology, ensuring the broadest possible coverage of this rapidly evolving PTM field.
Integrated Biological Context
Monoaminylation data is most powerful when interpreted within its biological framework. We provide optional bioinformatics integration with epigenetic pathway analysis, neurotransmitter metabolism context, and TGM2 expression data — enabling you to connect monoaminylation sites to their functional and regulatory environment. Our Rare PTM Library service provides complementary access to specialized methods for additional emerging and low-abundance modifications.
Publication-Ready Data
All deliverables are formatted to publication standards, including annotated spectra, quantitative tables, and methods sections ready for incorporation into manuscripts. We understand that monoaminylation data is often the first of its kind in a given biological system and provide the thorough documentation needed for high-impact publication.
Case Study: CRACR-Based Global Profiling of the Serotonylation Proteome in Cancer Cells
In a 2024 study published in Journal of Proteome Research (preprint: bioRxiv), Zhang et al. developed a pH-Controlled Chemoselective Rapid Azo-Coupling Reaction (CRACR) strategy for global profiling of the serotonylation proteome, dramatically expanding the known scope of this modification.
Background: Protein serotonylation — the TGM2-mediated covalent attachment of serotonin to glutamine residues — was known to occur on a limited number of substrates including small GTPases and histones. However, the full scope of the serotonylated proteome remained unknown due to the lack of efficient enrichment methods. Antibody-based approaches suffer from limited specificity and availability, while metabolic labeling methods require long incubation times and may alter cellular physiology.
Approach: The team designed a photoactive aryldiazonium-biotin probe that reacts selectively with serotonin moieties on proteins under mild, pH-controlled conditions. The CRACR method enables rapid (minutes), one-step labeling of serotonylated proteins directly in cell lysates without the need for metabolic incorporation. Labeled proteins are enriched via streptavidin-biotin affinity capture and identified by LC-MS/MS. The approach was validated in HCT 116 colorectal cancer cells, a cell line known to express high levels of TGM2.
Key Findings:
- Over 1,000 serotonylated proteins were identified — representing a >20-fold expansion over the previously known serotonylation proteome
- Serotonylated proteins span diverse functional categories including nucleic acid metabolism, RNA processing, chromatin regulation, cytoskeletal organization, and metabolic enzymes
- Multiple site-specific serotonylation events were mapped, enabling the first comprehensive view of serotonylation site preferences and sequence contexts
- Serotonylation levels were elevated in cancer cells compared to normal controls, consistent with TGM2 upregulation in malignancy
- The CRACR approach showed high specificity for serotonin over other biogenic amines and could be adapted for other monoaminylation types through probe design modifications
Significance: This study established that the serotonylated proteome is substantially larger than previously appreciated and provided a broadly applicable chemical tool for monoaminylation research. The CRACR method overcomes the long-standing enrichment bottleneck that has limited monoaminylation discovery and opens the door to systematic investigation of how serotonin signaling interfaces with the functional proteome in health and disease.
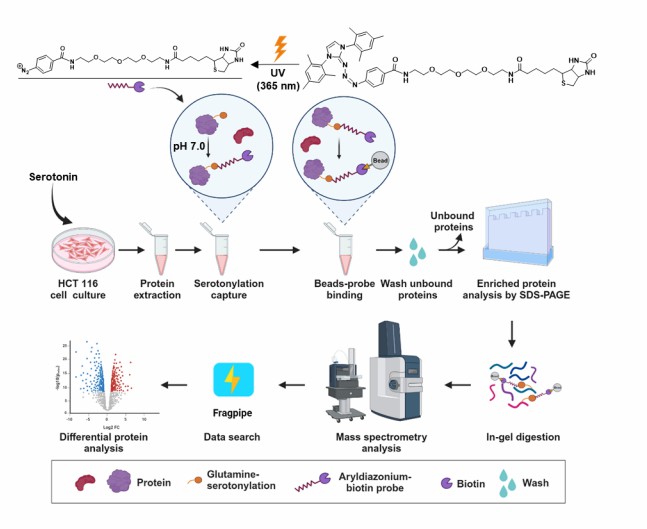
Figure 1 from Zhang et al. (2024). CRACR-based chemical proteomics strategy for global serotonylation profiling: pH-controlled chemoselective azo-coupling reaction, affinity enrichment, LC-MS/MS identification, and functional classification of the serotonylated proteome. (CC BY 4.0)
Representative Monoaminylation Analysis Results
Our monoaminylation analysis delivers integrated data packages with multiple annotation and visualization layers, providing comprehensive coverage of the monoaminylated proteome in your samples.
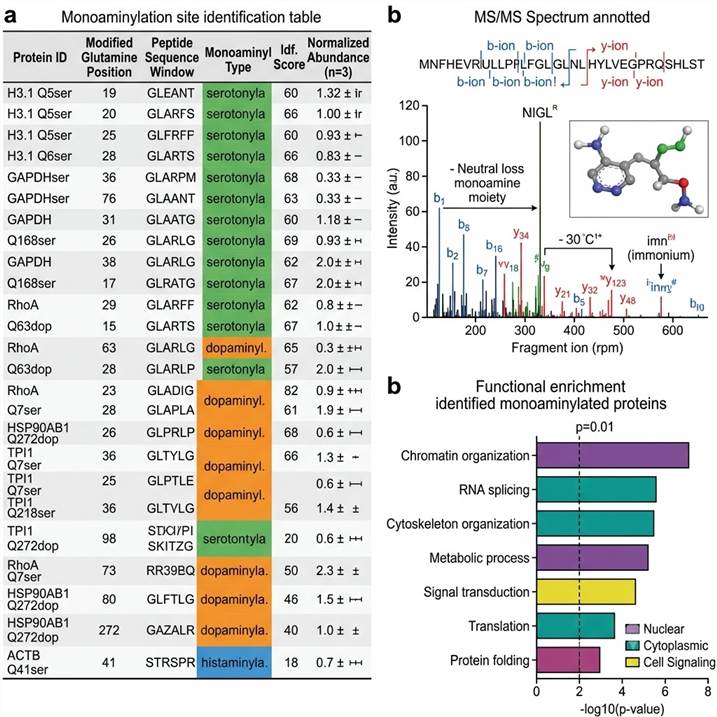
Representative data outputs from our protein monoaminylation analysis pipeline. Left: Monoaminylation site identification table. Center: Annotated MS/MS spectrum of a monoaminyl-modified peptide. Right: Functional enrichment analysis of monoaminylated proteins.
Key deliverables included in every project package:
- Monoaminylation site table — For each identified site: protein ID, modified glutamine position, peptide sequence, monoaminyl type (serotonyl/dopaminyl/histaminyl), identification confidence score, and spectral counts
- Annotated MS/MS spectra — Fragmentation spectra for each monoaminyl-modified peptide with diagnostic ions labeled, including the characteristic neutral loss signatures and immonium ions
- Quantitative comparison — For multi-condition experiments, site-specific monoaminylation abundance changes with statistical significance and effect sizes
- Functional enrichment analysis — GO term, pathway, and protein class enrichment for identified monoaminylated proteins, providing biological context for the modification landscape
- Labeling efficiency report — QC metrics for chemical probe labeling, enrichment efficiency, and replicate reproducibility
Related Services
Our monoaminylation analysis service is one component of a comprehensive emerging PTM and bioinformatics platform. These services can be used independently or integrated into a complete PTM discovery and characterization workflow.
FAQs
What is protein monoaminylation?
Protein monoaminylation is the covalent attachment of biogenic amine molecules — including serotonin (5-HT), dopamine, and histamine — to protein substrates, predominantly at glutamine residues. This reaction is catalyzed by transglutaminase 2 (TGM2) and results in an isopeptide bond between the amine and the glutamine side chain. Monoaminylation represents an emerging class of PTM that directly links neurotransmitter biology to protein function regulation.
What types of monoaminylation can you detect?
We offer detection and site mapping for protein serotonylation (serotonin attachment), dopaminylation (dopamine attachment), histaminylation (histamine attachment), and related biogenic amine modifications. Our chemical probe-based enrichment strategy can be adapted for additional monoamine substrates as needed for specific research applications.
Why is monoaminylation difficult to detect by standard proteomics?
Monoaminylation presents several analytical challenges: the isopeptide bond is acid-labile and can be lost under standard sample preparation conditions; there are no pan-specific antibodies available for enrichment of monoaminylated peptides; the modification occurs at low stoichiometry relative to unmodified protein; and the mass shift is relatively small and can be confused with other modifications without proper fragmentation evidence. Our chemical probe-based enrichment and specialized LC-MS/MS methods are specifically designed to overcome these challenges.
What is the biological significance of histone monoaminylation?
Histone monoaminylation at H3Q5 (serotonylation or dopaminylation) represents a direct link between neurotransmitter signaling and chromatin regulation. H3Q5ser promotes a permissive chromatin state and potentiates gene activation, while H3Q5dop has been implicated in cocaine-induced transcriptional plasticity. These marks coexist with adjacent H3K4me3 and add an additional layer of complexity to the histone code, with implications for understanding how neuronal activity translates into stable epigenetic changes.
What is the role of TGM2 in monoaminylation?
Transglutaminase 2 (TGM2) is the principal enzyme catalyzing protein monoaminylation. TGM2 crosslinks glutamine residues with primary amine substrates, and in the context of monoaminylation, it uses biogenic amines (serotonin, dopamine, histamine) as the amine donor. TGM2 expression is elevated in many cancer types, leading to increased monoaminylation levels in tumor tissues. TGM2 activity is regulated by calcium, GTP, and redox status, providing multiple points of regulation for monoaminylation levels in cells.
Can you quantify monoaminylation changes between conditions?
Yes. We offer both label-free quantification (based on extracted ion chromatogram comparison across runs) and TMT-based multiplexed quantification (up to 16 samples simultaneously). For specific monoaminylation sites of interest, we can also develop targeted PRM assays for precise, high-throughput quantification across many samples.
What controls are included in a monoaminylation experiment?
Every project includes: unmodified peptide controls for background subtraction, competition controls (free amine competition to validate labeling specificity), TGM2 inhibition controls where applicable, blank processing controls to identify probe-derived artifacts, and replicate injections for technical reproducibility assessment.
How does monoaminylation differ from other PTMs on glutamine residues?
Unlike the well-characterized glutamine deamidation (spontaneous conversion to glutamic acid) or methylation (methyl group addition), monoaminylation attaches a large, complex amine molecule (serotonin: 176 Da; dopamine: 153 Da) to the glutamine side chain via an isopeptide bond. This represents a substantial structural change that can affect protein-protein interactions, enzymatic activity, and subcellular localization. The TGM2-mediated mechanism is also shared with protein crosslinking, but monoaminylation results in stoichiometric modification rather than polymerization.
References
- Zhang N, Wu J, Gao S, Peng H, Li H, Gibson C, Wu S, Zhu J, Zheng Q. pH-Controlled Chemoselective Rapid Azo-Coupling Reaction (CRACR) Enables Global Profiling of Serotonylation Proteome in Cancer Cells. bioRxiv. 2024.05.10.593574.
- Zhang N, Gao S, Peng H, Wu J, Li H, Gibson C, Wu S, Zhu J, Zheng Q. Chemical Proteomic Profiling of Protein Dopaminylation in Colorectal Cancer Cells. bioRxiv. 2024.04.27.591460.
- Rangan RS, Petty R, Acharya S, Emmitte KA, do Valle RS, Lam C, Essajee SI, Mayhew W, Young O, Brooks CD, Forster MJ, Tovar-Vidales T, Clark AF. Phenethylaminylation: Preliminary In Vitro Evidence for the Covalent Transamidation of Psychedelic Phenethylamines to Glial Proteins. bioRxiv. 2025.02.13.638188.
For research use only. Not for use in diagnostic procedures.