Closing the Discovery-to-Validation Gap for Low-Abundance Modified Proteins
The fundamental challenge in PTM biomarker validation is not identification — modern phosphoproteomics routinely maps thousands of modification sites in a single experiment — but rather the ability to quantitatively verify specific low-abundance modification events across biologically relevant sample sets with sufficient sensitivity, specificity, and reproducibility. This gap is particularly acute for three common research scenarios:
- The academic researcher who has identified 50 significantly regulated phosphosites from a TMT-based phosphoproteomics screen of drug-treated versus control cells, but needs orthogonal MRM-based quantification of the top 10 candidates in independent biological replicates — and has only 50 µg of remaining protein lysate per sample
- The translational scientist who needs to quantify a specific phosphoprotein biomarker (e.g., phospho-STAT3 Y705, phospho-ERK T202/Y204) in 100 µL of plasma from a clinical cohort, where the target protein exists at low ng/mL concentrations against a mg/mL albumin background
- The pharmaceutical biomarker team measuring drug target engagement through phosphorylation of specific signaling node proteins in 2–5 mg core needle biopsies from a Phase I oncology trial, where sample irreplaceability demands maximum data yield from minimal material
Our ultra-sensitive platform directly addresses all three scenarios through a single integrated workflow. The table below maps the most common research objectives to our recommended technical approach, enabling you to quickly identify the optimal strategy for your specific validation needs.
Find Your Solution: Research Goal → Recommended Approach
| Your Research Goal |
Recommended Approach |
Key Techniques |
| Validate candidate phosphosites from global discovery phosphoproteomics in independent samples |
Targeted enrichment + nanoLC-MRM/PRM with stable isotope dilution |
TiO₂/IMAC phosphopeptide enrichment, scheduled MRM (QQQ) or PRM (Orbitrap), SIL-AQUA internal standards, transition optimization |
| Detect and quantify low-abundance phosphoproteins in plasma, serum, or CSF |
Immunoaffinity-enhanced targeted MS (SISCAPA + nanoLC-PRM) |
Anti-phosphopeptide antibody capture, magnetic bead immunoaffinity, nanoflow LC-PRM, stable isotope dilution quantification |
| Quantify drug target engagement biomarkers in limited biopsy material (1–10 mg tissue) |
Full-length protein IP + nanoLC-MS/MS |
Modification-specific or protein-specific antibody pulldown, on-bead digestion, high-resolution PRM, phosphosite stoichiometry calculation |
| Monitor modified protein biomarkers across longitudinal preclinical studies (50–500 samples) |
Multiplex MRM panel with batch QC workflow |
Scheduled MRM transitions (up to 50 targets/run), QConCAT internal standards, inter-batch normalization, QC sample framework |
| Confirm modification sites with very low stoichiometry (<1% occupancy) or confirm novel modification events |
Deep enrichment + high-sensitivity PRM with spectral validation |
Large-scale TiO₂/IMAC enrichment, offline fractionation, PRM with full MS/MS spectral confirmation, site occupancy calculation |
Three Complementary Platforms for Ultra-Sensitive Modified Protein Detection
Our service deploys three complementary detection and enrichment platforms, each optimized for specific combinations of sensitivity requirement, sample type, modification type, and research question. Platform selection — or combination — is guided by the specific characteristics of your target modifications, sample availability, and quantitative precision needs.
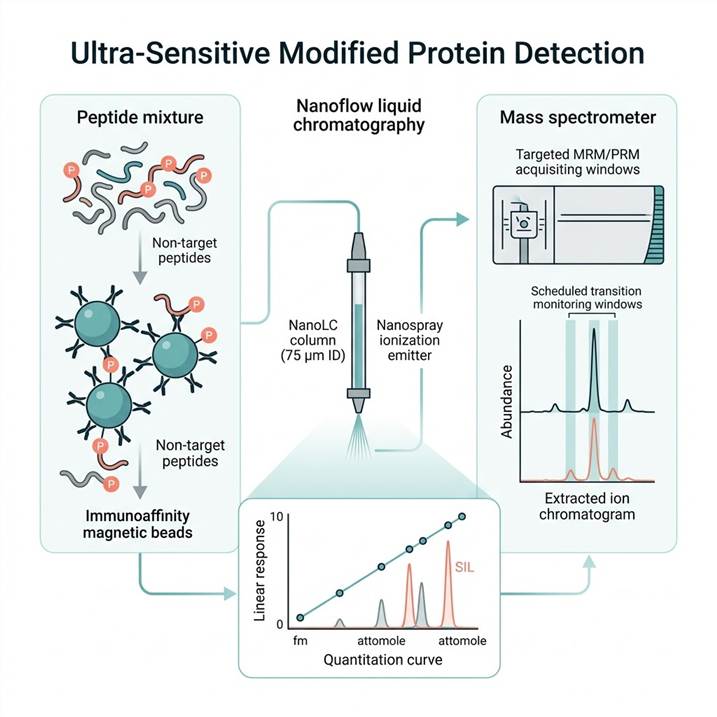
Nanoflow LC-MRM/PRM Targeted Mass Spectrometry
At the core of our ultra-sensitive detection capability is nanoflow liquid chromatography coupled to triple quadrupole (MRM) and high-resolution Orbitrap (PRM) mass spectrometers. NanoLC operation at 20–50 nL/min flow rates through 75 μm inner diameter analytical columns maximizes ionization efficiency and signal-to-noise ratios, delivering attomole-level limits of detection for modified peptides — approximately 100–1,000-fold improvement over conventional microflow LC-MS approaches. Scheduled MRM acquisition monitors both modified and unmodified peptide counterparts simultaneously within sub-minute retention time windows, enabling accurate stoichiometry determination for each target modification site. The PRM platform provides full MS/MS spectral acquisition across all target precursor ions, offering additional specificity through complete fragment ion series matching and high-resolution accurate mass measurement. This platform is optimal for projects with 5–50 predefined target modified peptides and is fully compatible with stable isotope-labeled internal standards (SIL-AQUA peptides at $500–$2,000 per target, or QConCAT proteins for larger panels at $5,000–$15,000 per construct).
Immunoaffinity-Enhanced Targeted MS (SISCAPA Workflow)
For the most demanding low-abundance applications — circulating phosphoprotein biomarkers in plasma, modified proteins in CSF, or rare modification events in limited biopsy material — the SISCAPA (Stable Isotope Standards and Capture by Anti-Peptide Antibodies) workflow provides unparalleled enrichment efficiency. Anti-peptide antibodies targeting specific modified sequences are covalently coupled to magnetic beads and used to capture target peptides (both modified and unmodified forms) from 100–500 µg of digested biological sample. This single-step immunoaffinity capture achieves up to 1,000-fold effective enrichment of target peptides, reducing the complex proteome background by 3–4 orders of magnitude before LC-MS analysis. Following magnetic bead separation and elution, captured peptides are quantified by nanoflow LC-MRM/PRM with SIL internal standards for absolute quantification. The SISCAPA workflow is particularly powerful for clinical matrix applications where the combination of limited sample volume (100–500 µL plasma), complex protein background (40–60 mg/mL total protein), and low target abundance (low fmol/mg levels) demands maximum enrichment prior to MS analysis. For projects requiring standalone enrichment method development, our Modified Peptide Enrichment Services provide a comprehensive menu of enrichment options for different stages of the discovery-validation pipeline.
Full-Length Modified Protein Immunoaffinity Capture with MS Readout
When the biological target is a specific modified protein rather than isolated modification sites, whole-protein immunoaffinity capture provides a fundamentally different enrichment strategy with distinct advantages. Target proteins are immunoprecipitated from native or denatured lysates using modification-specific antibodies (e.g., anti-phosphotyrosine, anti-acetyllysine) or protein-specific antibodies, followed by on-bead digestion and LC-MS analysis. This approach preserves the native protein context of all co-occurring modifications on the same molecule, reduces sample complexity by 2–3 orders of magnitude before digestion, and enables simultaneous monitoring of multiple modification sites on the same target protein in a single analysis. It is particularly valuable for studies of phosphorylation signaling networks where the activation state of specific pathway nodes — defined by the occupancy of multiple phosphorylation sites on key signaling proteins such as kinases, adaptors, and transcription factors — must be assessed from limited biological material. When paired with our PTM Site Occupancy Analysis service, this approach delivers comprehensive site-level stoichiometry data integrating both modified and unmodified peptide quantification across the target protein.
Why Choose Our Ultra-Sensitive Modified Protein Detection Service
Attomole-Level Sensitivity from Minimal Starting Material
Nanoflow LC-MS operation at 20 nL/min provides 100–1,000-fold higher ionization efficiency than conventional microflow LC-MS, enabling confident modified peptide detection from as little as 1 µg of peptide digest. When combined with immunoaffinity enrichment, this platform achieves limits of quantification at low attomole on-column — representing modified protein concentrations in the low fmol/mg range in the original biological sample.
Orthogonal Validation from Discovery Through Verification
Unlike immunoassay-only approaches that depend entirely on antibody availability and specificity, our MS-based targeted platform provides orthogonal validation with independent detection chemistry. MRM/PRM targets are defined by peptide sequence, modification mass shift, and fragmentation pattern — not by antibody epitope accessibility — providing an orthogonal measurement dimension that strengthens the overall validation chain from discovery to biological confirmation.
Absolute Quantification Across Study Cohorts and Time
Stable isotope-labeled internal standards spiked at known concentrations into every sample enable absolute quantification (reported in fmol/mg total protein or amol/µg digest), not relative abundance ratios. This absolute quantification framework supports cross-study comparison, inter-laboratory data sharing, and longitudinal biomarker monitoring where batch-to-batch consistency is essential for meaningful biological interpretation.
Integrated PTM Analysis Ecosystem
Our ultra-sensitive detection platform is embedded within a comprehensive PTM analysis infrastructure spanning global discovery (Global PTM Profiling), targeted verification (PRM PTM Verification, this service), antibody-based screening (PTM Antibody Array, Multiplex PTM Immunoassay), and bioinformatics integration (PTM Bioinformatics Analysis). Discovery findings identified through global profiling can be transitioned directly to targeted assay development within a single service organization, preserving experimental continuity and accelerating the discovery-to-validation timeline.
Workflow: From Candidate Target to Validated Modified Protein Quantification
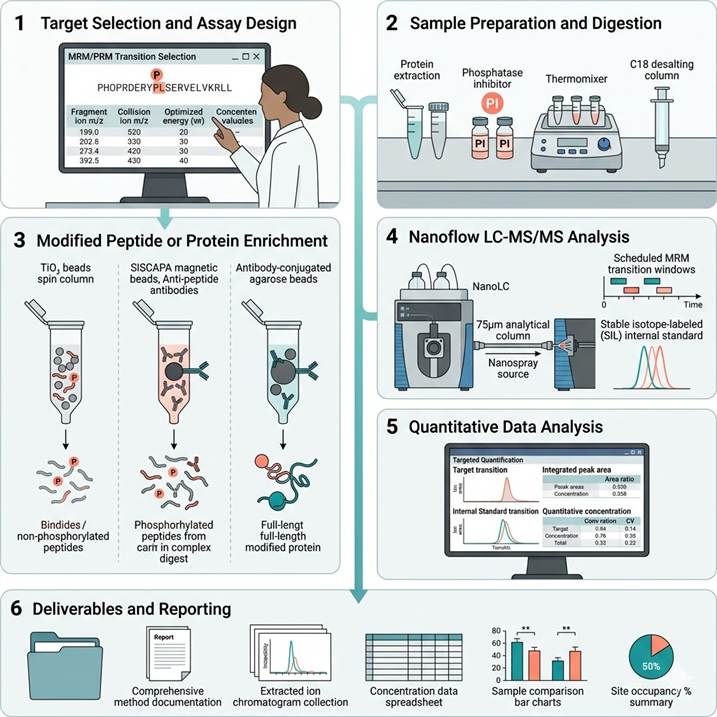
Step 1: Target Selection and Assay Design
We collaborate with you to define target modified proteins or specific modification sites, design MRM/PRM methods (transition selection, collision energy optimization, retention time scheduling), and select the optimal enrichment strategy based on modification type, sample matrix, target abundance, and quantitative precision requirements.
Step 2: Sample Preparation and Digestion
Protein extraction, reduction, alkylation, and enzymatic digestion are performed using protocols optimized for modified peptide recovery. For phosphoprotein analysis, phosphatase inhibitors are included throughout processing to preserve modification status. Optional peptide fractionation can be incorporated for improved detection depth in complex samples.
Step 3: Modified Peptide or Protein Enrichment
Targeted enrichment is performed using the selected strategy: TiO₂/IMAC for global phosphopeptide enrichment, SISCAPA immunoaffinity beads for specific modified peptide capture with up to 1,000-fold enrichment, or antibody-based pulldown for full-length modified protein enrichment. Enrichment efficiency is monitored at each stage.
Step 4: Nanoflow LC-MS/MS Analysis
Enriched samples are analyzed by nanoflow LC-MS/MS with targeted MRM or PRM acquisition at 20–50 nL/min flow rates. Stable isotope-labeled internal standard peptides (SIL-AQUA or QConCAT) are spiked at known concentrations for absolute quantification. Scheduled retention time windows ensure optimal MS duty cycle for each target.
Step 5: Quantitative Data Analysis
Raw LC-MS data are processed using targeted quantification software for peak detection, integration, and isotope ratio calculation. Transition chromatograms are manually reviewed for interference assessment. Absolute concentrations are calculated from SIL internal standard response, and modification site occupancy is determined from paired modified/unmodified peptide ratios.
Step 6: Deliverables and Reporting
Complete quantitative report including: MRM/PRM method parameters and transition details, extracted ion chromatograms for each target with internal standard overlay, calculated absolute concentrations with statistical metrics, site occupancy determinations where applicable, and individual sample value plots with group comparison statistics.
Applications in Low-Abundance Modified Protein and Biomarker Research
Our ultra-sensitive modified protein detection platform supports five principal application areas where the combination of low target abundance, limited sample availability, and high quantitative precision demands the sensitivity and specificity that only immunoaffinity-enhanced nanoflow targeted MS can provide.
Phosphoprotein Biomarker Validation from Discovery Cohorts
Discovery phosphoproteomics using TMT, label-free, or DIA approaches identifies hundreds to thousands of differentially phosphorylated sites, but biological validation requires orthogonal, quantitative measurement of these candidates in independent sample cohorts. Our service provides the sensitivity to quantify even low-abundance phosphoprotein biomarkers from limited clinical or preclinical sample sets, with throughput sufficient for cohort-scale validation (20–200 samples per study). Typical project scope: 5–20 target phosphosites, 50–100 µg starting protein per sample, 3–6 week turnaround for a 50-sample cohort. For larger cohort screening needs, our Multiplex PTM Immunoassay Services provide complementary higher-throughput antibody-based detection.
Low-Stoichiometry PTM Confirmation (<1% Occupancy)
Many biologically critical modification events — particularly phosphorylation of transcription factors (e.g., phospho-p53 S15, phospho-STAT3 Y705), signaling adaptors, and scaffolding proteins — occur at very low stoichiometry that falls below the detection threshold of conventional targeted MS approaches. Our workflow, combining deep enrichment with nanoflow LC-PRM, routinely quantifies modification sites at <1% occupancy from 100–500 µg of protein lysate. This capability is essential for confirming modification events that are identified in discovery datasets but cannot be validated by standard MRM. For broader coverage of low-abundance PTM sites, our Label-Free PTM Quantification and Global PTM Profiling services provide complementary discovery-phase approaches.
Drug Target Engagement via Phosphoprotein Signaling Analysis
In drug development programs, direct measurement of specific phosphorylation events on kinase substrates, signaling node proteins, and downstream effectors provides the most direct evidence of intracellular target engagement and pathway modulation. Our ultra-sensitive platform enables quantification of these phosphorylation events from limited clinical material — core needle biopsies (1–5 mg), fine-needle aspirates, or laser-capture microdissected tissue — that are inaccessible to conventional MS-based phosphoproteomics. This capability is particularly valuable for early-phase clinical trials where pharmacodynamic biomarker assessment in on-treatment tumor biopsies is a critical proof-of-mechanism endpoint. For broader kinome-level signaling analysis, our Kinase-Substrate Network Analysis service provides complementary pathway-level phosphoproteomic profiling.
Clinical Matrix Modified Protein Detection (Plasma, Serum, CSF)
Clinically accessible biofluids contain modified proteins at extremely low absolute concentrations — typically low pg/mL to ng/mL — embedded within a high-abundance protein background (40–60 mg/mL in plasma, 15–45 mg/mL in CSF). Immunoaffinity-enhanced targeted MS (SISCAPA) is uniquely suited to this analytical challenge: anti-peptide antibodies provide the specificity to extract and enrich specific modified peptides from complex clinical matrices, while nanoflow LC-MRM/PRM delivers the sensitivity to quantify them at clinically relevant concentrations from 100–500 µL of biofluid. This application is directly relevant to circulating phosphoprotein biomarker development, liquid biopsy approaches targeting modified proteins, and longitudinal monitoring of protein modification biomarkers in clinical sample collections.
Preclinical Modified Protein Biomarker Monitoring
Longitudinal preclinical studies — including xenograft models, transgenic mouse studies, and non-human primate pharmacology studies — generate valuable sample series where modified protein biomarker levels must be quantified across multiple timepoints, dose groups, and treatment conditions. Our targeted MRM/PRM platform provides the quantitative consistency required for robust temporal profiling, with inter-batch CVs typically <15% when using stable isotope-labeled internal standards and standardized QC samples. Typical project scope: 5–15 target modified peptides, 50–200 timepoint samples, 4–8 week turnaround with interim data reviews at protocol-defined milestones.
Representative Results: Performance Data and Project Deliverables
Our ultra-sensitive modified protein detection platform delivers quantitative data packages designed to support both publication-grade reporting and regulatory-compliant biomarker documentation. The representative data below illustrates the typical analytical performance achieved across different sample types and target modification classes.
Typical Assay Performance Specifications
| Performance Parameter |
Direct MRM/PRM (no enrichment) |
TiO₂/IMAC Enriched |
SISCAPA Immunoaffinity |
Whole-Protein IP |
| On-column LOD (modified peptide) |
5–50 amol |
1–10 amol |
0.5–5 amol |
2–20 amol |
| Typical starting material |
50–200 µg protein |
200–1000 µg protein |
100–500 µg digest |
200–1000 µg protein |
| LOQ in original sample |
10–100 fmol/mg |
1–10 fmol/mg |
0.5–5 fmol/mg |
2–20 fmol/mg |
| Inter-assay CV |
<10% |
<15% |
<15% |
<20% |
| Linear dynamic range |
3–4 orders |
2–3 orders |
2–3 orders |
2–3 orders |
| Throughput (samples/week) |
50–100 |
20–50 |
20–40 |
15–30 |
| Multiplexing capacity (targets/run) |
10–50 |
5–20 |
1–10 |
1–5 |
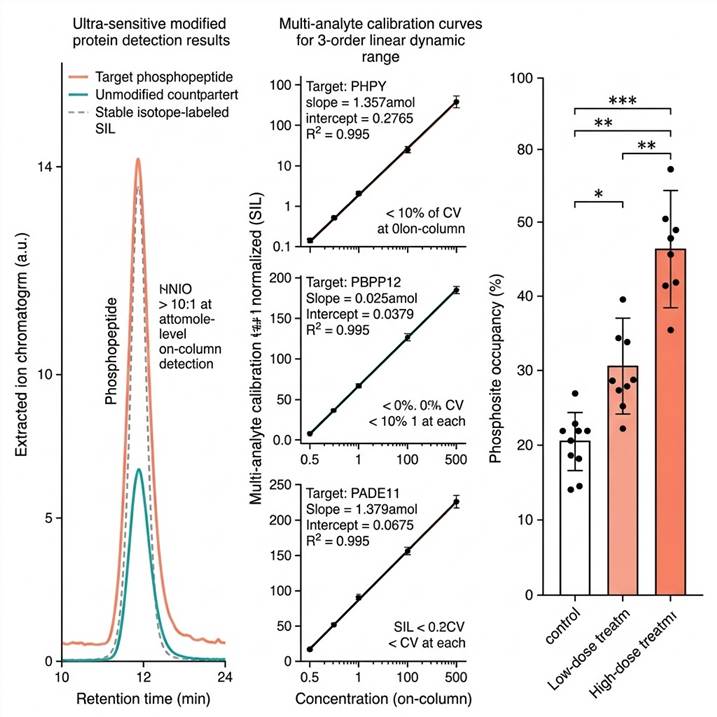
Representative data outputs from our Ultra-Sensitive Modified Protein Detection platform. Left: Extracted ion chromatograms with SIL internal standard. Center: Calibration curves and linear dynamic range. Right: Quantitative group comparison with statistical analysis.
Key quantitative deliverables included in every ultra-sensitive modified protein detection project:
- Method documentation package — Complete MRM/PRM method parameters including peptide sequences, precursor/product ion m/z values, collision energies, retention time windows, and internal standard concentrations for each target
- Extracted ion chromatograms — Publication-ready EICs for each target peptide with SIL internal standard overlay, retention time alignment verification, and signal-to-noise ratio reporting for quality assessment
- Quantitative concentration data — Absolute or relative concentrations for each target in each sample, calculated from SIL internal standard response, with CV and confidence intervals
- Site occupancy analysis — Occupancy percentages for each modification site calculated from paired modified/unmodified peptide ratios where both forms are detectable, with occupancy confidence intervals
- Statistical analysis — Group comparison statistics including fold-change, p-values (t-test, ANOVA, or non-parametric tests as appropriate), and individual value plots with group means for each target across all experimental groups
- Data visualization — Publication-quality bar charts, box plots, or heat maps summarizing quantitative results across all targets and sample groups, formatted for direct use in manuscripts and presentations
Related Services
Our ultra-sensitive modified protein detection platform is part of a comprehensive PTM analysis service portfolio spanning targeted MS, immunoassay, and bioinformatics solutions for integrated PTM research from candidate discovery through biological validation.
- PRM PTM Verification — Parallel reaction monitoring-based targeted verification of PTM sites with full MS/MS spectral confirmation and high-resolution accurate mass quantification
- Modified Peptide Absolute Quantification — Absolute quantification of modified peptides using stable isotope-labeled internal standards with MRM and PRM acquisition for precise concentration determination
- Modified Peptide Enrichment Services — Comprehensive enrichment solutions including IMAC, TiO₂, HILIC, and antibody-based immunoaffinity approaches for modified peptide isolation prior to MS analysis
- PTM Site Occupancy Analysis — Dedicated site occupancy determination service quantifying the proportion of modified versus unmodified protein at specific modification sites using targeted MS approaches
- Multiplex PTM Immunoassay Services — High-throughput Luminex, ECL, and ELISA-based multiplexed PTM detection for large-cohort biomarker validation and drug development studies
- PTM Bioinformatics Analysis — Advanced bioinformatics analysis of PTM datasets including pathway enrichment, modification crosstalk, motif analysis, and multi-omics data integration
FAQs
What is the sensitivity limit of this ultra-sensitive modified protein detection service?
For optimized MRM/PRM assays with nanoflow LC-MS, we routinely achieve 5–50 attomole limits of detection on-column for modified peptides without enrichment. With immunoaffinity enrichment (SISCAPA), detection sensitivity extends to 0.5–5 attomole on-column, enabling quantification of modified peptides present at 0.5–5 fmol/mg total protein in the original biological sample. Actual sensitivity depends on target peptide ionization efficiency, modification type (phosphorylation reduces ionization ~2–5-fold relative to unmodified), sample matrix complexity, and enrichment efficiency.
What amount of starting material is required for this service?
Sample requirements depend on the chosen enrichment strategy and target abundance. For direct MRM/PRM assays without immunoaffinity enrichment, 50–200 µg of total protein is recommended. With TiO₂/IMAC enrichment, 200–1000 µg of protein is typically used. With SISCAPA immunoaffinity enrichment, 100–500 µg of total protein digest is sufficient. For the most precious samples — laser capture microdissected tissue, FACS-sorted cell populations, or clinical biopsies — optimized nanoflow workflows can work with as little as 1–10 µg of peptide digest, with appropriate adjustments to detection limits.
How does this ultra-sensitive detection service differ from global PTM discovery services?
Global PTM discovery services prioritize breadth of coverage — maximizing the number of identified modification sites across the proteome — and are typically performed using data-dependent acquisition (DDA) or data-independent acquisition (DIA) on bulk samples. Our ultra-sensitive targeted detection service prioritizes sensitivity, specificity, and quantitative accuracy for a predefined set of target modification sites, using targeted acquisition modes (MRM/PRM) with stable isotope-labeled internal standards. It is designed for verification and validation of specific candidate modification sites, not for broad discovery. A typical use case: a researcher identifies 50 regulated phosphosites by DIA phosphoproteomics (discovery), then selects the top 10 for targeted MRM validation in 100 independent samples using our service (verification).
Can you develop custom targeted assays for my specific modified proteins of interest?
Yes. For each target modification site, we develop, optimize, and validate a custom targeted MS method. This process includes selection of proteotypic peptides covering the modification site, MRM/PRM transition design (typically 3–5 transitions per precursor) and collision energy optimization, stable isotope-labeled internal standard synthesis (SIL-AQUA peptides at $500–$2,000 per target, or QConCAT proteins at $5,000–$15,000 for larger panels), enrichment protocol optimization for the specific modification type, and analytical validation including linearity assessment (minimum 3 orders of dynamic range), precision determination (intra- and inter-assay CV), and recovery/spike-recovery evaluation.
What types of post-translational modifications can be detected with this service?
Our service is optimized for phosphorylation (p-Ser, p-Thr, p-Tyr) detection and quantification, which represents the primary application (>80% of projects). We also support targeted detection and quantification of ubiquitination (di-Gly remnant peptides, +114.04 Da mass shift on Lys), acetylation (+42.01 Da on Lys), methylation (mono- +14.02 Da, di- +28.03 Da, tri- +42.05 Da on Lys or Arg), SUMOylation (remnant peptide signature), and other PTMs where the modification produces a detectable and fragmentation-stable mass shift. Method sensitivity is optimized for each modification type based on enrichment efficiency, ionization characteristics, and fragmentation behavior. Enrichment strategy and LC-MS parameters are adjusted per modification type to achieve optimal performance.
Can this service be applied to clinical sample matrices such as plasma or CSF?
Yes — clinical matrix analysis is a core application of our immunoaffinity-enhanced targeted MS (SISCAPA) platform. We have extensive experience detecting and quantifying low-abundance modified proteins in plasma (typically 100–500 µL per assay), serum, CSF (1–5 mL), tissue biopsies (1–10 mg), and other clinically relevant matrices. The SISCAPA approach is particularly effective for clinical samples, where anti-peptide antibodies provide the specificity to extract target modified peptides from the complex protein background of biofluids (40–60 mg/mL total protein in plasma) while nanoflow LC-MRM/PRM delivers the sensitivity needed for clinical-level quantification. Matrix-specific validation — including spike recovery assessment, matrix effect evaluation, and dilution linearity testing — is performed for each new matrix type.
What quantitative data and deliverables are included in the final report?
Each project delivers a comprehensive quantitative report including: (1) complete MRM/PRM method parameters and transition details for each target peptide; (2) extracted ion chromatograms for all target peptides with stable isotope-labeled internal standard overlay and retention time alignment verification; (3) calculated absolute or relative concentrations with statistical metrics (CV, standard deviation, 95% confidence intervals); (4) modification site occupancy percentages where paired modified/unmodified peptide data are available, with occupancy confidence intervals; (5) individual sample value plots with group comparison statistics (fold change, p-values from appropriate statistical tests); (6) detailed methods documentation suitable for publication and regulatory reference including sample preparation protocols, enrichment procedures, LC-MS parameters, and data analysis workflows. Raw data files (RAW format) are provided upon request for independent re-analysis.
References
- Dakup PP, Feng S, Shi T, Jacobs JM, Wiley HS, Qian WJ. Targeted quantification of protein phosphorylation and its contributions towards mathematical modeling of signaling pathways. Molecules. 2023;28(3):1143.
- Whiteaker JR, Zhao L, Kennedy JJ, Ivey RG, Paulovich AG. Targeted mass spectrometry for quantification of receptor tyrosine kinase signaling. In: Proteomics for Drug Discovery. Methods in Molecular Biology, Vol. 2823. Springer; 2024:253-267.
- Babaei M, Kashanian S, Lee HT, Harding F. Proteomics techniques in protein biomarker discovery. Quantitative Biology. 2024;12(1):53-69.
For research use only. Not for use in diagnostic procedures.