Optimizing Sample Preparation for DIA Proteomics: Best Practices
Data-Independent Acquisition (DIA) is transforming the landscape of proteomics by enabling high-throughput, comprehensive, and reproducible protein quantification across complex biological samples. Unlike Data-Dependent Acquisition (DDA), DIA fragments all peptides within predefined m/z windows, providing a systematic and unbiased approach for deep proteome coverage. This unique advantage makes DIA particularly valuable in large-scale biomarker discovery, drug development pipelines, and systems biology studies.
However, the power of DIA relies heavily on sample quality. Poorly prepared samples lead to ion suppression, missed cleavages, and incomplete proteome coverage, resulting in low reproducibility and compromised quantitative accuracy. Unlike targeted or DDA-based workflows, DIA's global acquisition strategy amplifies the consequences of suboptimal sample handling because any impurity or technical variability affects the entire dataset.
This article provides an end-to-end guide to optimizing sample preparation for DIA proteomics, covering critical steps, troubleshooting strategies, and advanced methods for handling complex matrices and scaling high-throughput projects.
Why Sample Preparation Quality Determines DIA Success
Impact on Quantitative Accuracy and Proteome Coverage
DIA quantification depends on consistent fragment ion intensity across multiple windows and replicates. Even small variations in sample composition can lead to significant differences in precursor detection and peptide scoring.
- Quantitative implications: Poor digestion efficiency or incomplete solubilization can result in peptide losses that bias statistical analyses. For large projects, maintaining CV <15% across biological replicates is a widely accepted standard for DIA reproducibility.
- Proteome coverage trade-off: High-abundance contaminants, salts, and detergents interfere with ionization, reducing MS sensitivity and preventing low-abundance proteins from being detected.
Challenges Unique to DIA Sample Prep
Compared with DDA, DIA covers the full precursor ion range in every cycle, making the signal-to-noise ratio (SNR) highly sensitive to contaminants and non-protein components. Common issues include:
- Residual detergents (e.g., SDS) causing severe ion suppression.
- Batch effects due to inconsistent digestion conditions or peptide cleanup methods.
- Variable protein input across samples leading to skewed normalization during DIA-NN or Spectronaut analysis.
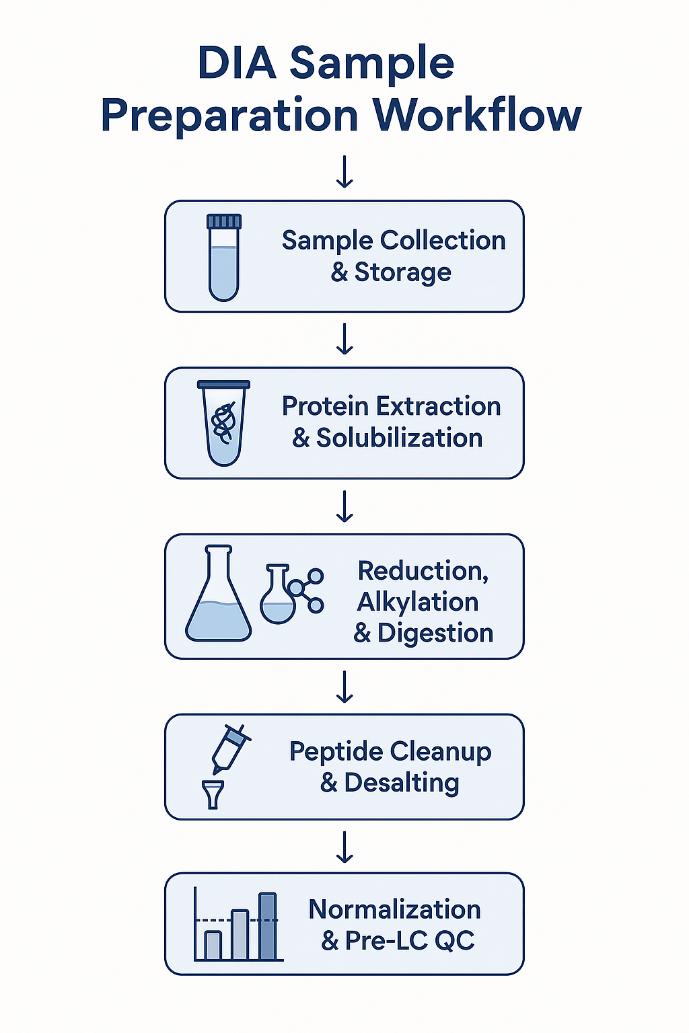
Key Steps in DIA Sample Preparation and Their Optimization
Sample Collection and Storage
High-quality DIA proteomics starts with standardized sample collection. Inconsistent handling introduces batch effects and protein degradation that cannot be corrected downstream.
Best Practices:
- Containers: Use low-binding polypropylene tubes to prevent peptide adsorption, which can reduce peptide recovery by up to 15%.
- Temperature Control: Immediately snap-freeze samples in liquid nitrogen and store at -80°C. Avoid more than one freeze–thaw cycle, as repeated cycles increase protein degradation and PTM loss.
- Matrix-Specific Considerations:
- Plasma/Serum: Implement high-abundance protein depletion (albumin, IgG) to improve dynamic range by up to 2–3×. For deeper profiling, extracellular vesicle (EV) removal reduces background interference.
- Tissue Samples: Maintain consistent homogenization speed and buffer composition. Add protease and phosphatase inhibitors during lysis to preserve post-translational modifications critical for biomarker discovery.
QC Tip:
Include a pooled QC sample representing all experimental groups to monitor variance during DIA acquisition and data processing.
Protein Extraction and Solubilization
Effective lysis and solubilization are essential for maximizing protein recovery, especially in complex matrices like tissues and biofluids.
Recommended Buffers:
- Chaotropic Agents: Urea (6–8 M) or thiourea efficiently disrupt protein aggregates and are MS-compatible after dilution.
- MS-Compatible Surfactants: Sodium deoxycholate (SDC) and RapiGest enable strong solubilization but can be easily removed during peptide cleanup.
- Avoid SDS unless necessary: While SDS is a powerful solubilizer, residual SDS causes up to 90% ion suppression in LC-MS. If used, employ FASP (Filter-Aided Sample Preparation) for removal.
Mechanical Disruption:
- Apply sonication or bead milling, but maintain consistent cycles to prevent heat-induced degradation.
- Excessive sonication can oxidize methionine residues, introducing artificial modifications.
QC Tip:
- Quantify protein concentration (BCA or Bradford assay) to normalize sample input. Aim for ≥50 µg total protein per sample for robust DIA quantification.
Reduction, Alkylation, and Digestion
Enzymatic digestion is critical for generating peptides with suitable charge states and lengths for DIA analysis.
Reduction & Alkylation:
- Reduce disulfide bonds with DTT (5–10 mM) or TCEP at 37°C for 30–60 minutes.
- Alkylate cysteines using iodoacetamide (IAA, 10–20 mM) in the dark to prevent reformation of disulfides.
- Warning: Overalkylation or carbamylation can introduce artifactual modifications, leading to false positives in PTM analysis. Maintain pH at 8.0 and avoid high temperatures (>37°C).
Protease Selection:
- Trypsin remains the gold standard, cleaving after Lys/Arg for predictable peptide lengths.
- For challenging matrices, a dual-enzyme approach (Trypsin + Lys-C) increases cleavage efficiency and reduces missed cleavages.
Digestion Conditions:
- Enzyme-to-protein ratio: 1:50–1:100.
- Incubation: 12–16 hours at 37°C with gentle agitation.
- QC Check: Missed cleavage rate should be <15%, assessed by a pilot nanoLC-MS run or SDS-PAGE.
Peptide Cleanup and Desalting
Post-digestion cleanup is critical for removing salts and detergents that impair LC-MS performance.
Common Methods:
- Solid-Phase Extraction (SPE): C18 cartridges provide high peptide recovery (>80%) and salt removal.
- StageTips: Ideal for low-input samples and compatible with automation.
- Mixed-Mode SPE: Useful for removing residual surfactants such as SDC.
Incomplete desalting leads to ion suppression and column clogging, significantly reducing the sensitivity and reproducibility of DIA runs.
QC Tip:
Measure peptide recovery post-cleanup using UV absorbance at 214 nm or 280 nm to ensure consistency across batches.
Sample Normalization and Pre-LC QC
Unequal peptide loading can cause major batch effects in DIA analysis, even with advanced normalization algorithms like those in Spectronaut or DIA-NN.
Normalization Practices:
- Adjust peptide concentration to 0.5–1 µg/µL in 0.1% FA before injection.
- Spike-in iRT (indexed Retention Time) peptides for retention time calibration and LC-MS performance monitoring.
- Pool technical replicates to create a QC sample, which should be analyzed at regular intervals during the LC-MS sequence to detect instrument drift.
QC Metrics Before Run:
- Peptide concentration CV should be <10% across samples.
- Verify sample clarity (no visible particulates) to prevent clogging nanoLC columns.
Common Pitfalls in DIA Sample Preparation and How to Avoid Them
Suboptimal sample preparation remains one of the most frequent reasons for DIA proteomics project failure. These issues often originate during collection, digestion, or cleanup steps and can propagate through the entire workflow, impacting both quantitative accuracy and proteome coverage. Below is a detailed guide to the most common pitfalls, their consequences, and practical strategies for prevention.
| Pitfall | Impact on DIA Data | Root Cause | Preventive Action & QC Checks |
| Incomplete Digestion |
|
|
|
| Detergent Contamination |
|
|
|
| Overalkylation / Carbamylation |
|
|
|
| Batch Inconsistency |
|
|
|
| Salt Carryover |
|
|
|
| Protein Loss During Cleanup |
|
|
|
Advanced Strategies for High-Throughput and Complex Samples
- Automated platforms: Robotic sample prep systems reduce human error and batch effects in large-scale DIA studies.
- TMT-DIA hybrid workflows: Combining multiplexed TMT labeling with DIA increases throughput and improves statistical power for multi-group comparisons.
- EV-depletion in plasma workflows: Enhances sensitivity for low-abundance biomarkers.
Quality Control Checkpoints Before DIA Run
Quality control (QC) should be embedded throughout the sample preparation process, not as an afterthought. QC checkpoints ensure data quality and reproducibility by identifying issues before LC-MS injection.
Pre-Digestion QC
Protein Quantification:
- Target: ≥50 µg per sample for standard DIA.
- Method: BCA or Bradford assay.
Homogeneity Check:
- Visual inspection for particulates; centrifuge if needed.
Post-Digestion QC
Peptide Recovery Rate:
- Target: ≥80% recovery compared to theoretical yield.
- Method: UV absorbance at 214 nm or NanoDrop.
Missed Cleavage Rate:
- Acceptable threshold: <15%.
- Method: Quick LC-MS pilot run or DIA-NN digestion stats.
Cleanup Efficiency QC
Detergent Removal: SDS level should be below detection threshold (colorimetric assay or test run).
Salt Carryover: Check conductivity of eluate or monitor MS baseline noise.
Pre-LC-MS QC
Peptide Load Normalization: Target: 0.5–1 µg/µL in 0.1% FA.
System Suitability Test: Inject iRT peptide mix to verify retention time reproducibility (R² ≥ 0.98).
QC Sample Analysis: Analyze pooled QC after every 5–10 injections; monitor TIC stability and PCA clustering.
Suggested QC Table for Web Integration
| QC Step | Metric | Acceptance Criteria |
| Protein Input | Concentration | ≥50 µg per sample |
| Peptide Recovery | Yield after cleanup | ≥80% |
| Missed Cleavages | DIA-NN stats | <15% |
| iRT Correlation | Retention time reproducibility | ≥0.98 |
| CV Across Samples | Technical replicates | <15% |
References:
- Zhang, Fangfei, et al. "Data‐independent acquisition mass spectrometry‐based proteomics and software tools: a glimpse in 2020." Proteomics 20.17-18 (2020): 1900276.
- Ludwig, Christian, et al. "Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial." Molecular Systems Biology 14.8 (2018): e8126.
- Bruderer, Roland, et al. "Analysis of 1508 plasma samples by capillary-flow data-independent acquisition profiles proteomics of weight loss and maintenance." Molecular & Cellular Proteomics 18.6 (2019): 1242-1254.
- Bekker-Jensen, Daniel B., et al. "A compact quadrupole-orbitrap mass spectrometer with FAIMS interface improves proteome coverage in short LC gradients." Molecular & Cellular Proteomics 19.4 (2020): 716-729. DOI: 10.1074/mcp.TIR119.001906
- Doellinger, Joern, et al. "Sample preparation by easy extraction and digestion (SPEED)–a universal, rapid, and detergent-free protocol for proteomics based on acid extraction." Molecular & Cellular Proteomics 19.1 (2020): 209-222. DOI: 10.1074/mcp.TIR119.001616
- Reiter, Lukas, et al. "Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry." Molecular & Cellular Proteomics 8.11 (2009): 2405-2417. DOI: 10.1074/mcp.M900317-MCP200

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)