Phosphoproteomics Service
- Service Details
- Case Study
In the field of life sciences research, the study of proteins and their post-translational modifications (PTM) is crucial for understanding the complex molecular mechanisms behind various cellular processes. Creative Proteomics based on data-independent acquisition (DIA) for phosphoproteomics analysis, can help researchers delve deeper into the complexity of the proteome. Our service provides effective technical support for phosphoproteomics analysis and plays an important role in helping researchers advance the field of DIA-based proteomics research.
DIA Proteome Analysis
DIA proteome analysis is a robust and versatile method for the comprehensive study of the proteome within biological samples. It involves the systematic identification and quantification of a wide range of proteins present in a given sample. DIA does not rely on precursor ion selection, making it highly suitable for quantitative proteome analysis. This service enables researchers to gain a holistic view of the proteome's composition, facilitating the discovery of biomarkers, elucidation of cellular pathways, and in-depth characterization of protein profiles.
Phosphoproteomics Analysis
Phosphorylation is undoubtedly the most important form of post-translational modification ubiquitous in prokaryotic and eukaryotic cells, which can lead to changes in protein structure, activation/inhibition of protein activity, and promotion/prevention of protein-protein interactions. Phosphorylation is at the core of many biological processes, including the regulation of signal transduction, gene expression and cell division. In humans, aberrant phosphorylation of proteins is commonly associated with a number of diseases such as cancer[1]. Therefore, a better understanding of protein phosphorylation will help deepen our understanding of important cellular processes, better understand disease mechanisms, and drive the discovery of effective therapies and new biomarkers.
DIA phosphoproteomics analysis is an extension of DIA proteome analysis specifically tailored to the comprehensive study of phosphorylated proteins and their PTMs. By combining the power of DIA with phosphopeptide enrichment techniques, this service allows for the systematic identification and quantification of phosphorylated proteins in complex biological samples. This in-depth analysis is crucial for unraveling the intricacies of signaling pathways, drug development, and biomarker discovery in the context of diseases.
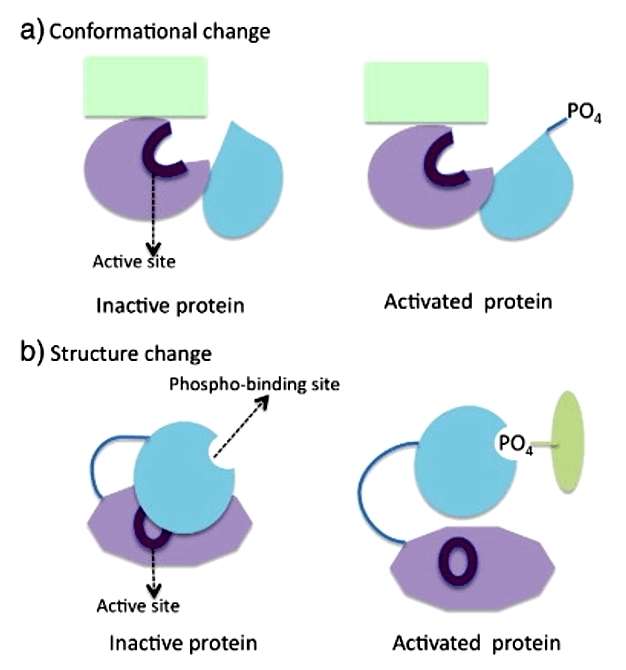
Technical Approach of Phosphoproteomics
DIA proteome analysis relies on a data-independent approach, wherein all precursor ions within a specified mass-to-charge (m/z) range are fragmented in a systematic manner. This fragmentation produces a series of fragment ions, enabling the accurate identification and quantification of peptides and proteins. The method typically involves liquid chromatography-mass spectrometry (LC-MS/MS) to separate and analyze peptides, followed by database searches for peptide identification. The use of stable isotope labeling or label-free quantification techniques further enhances the accuracy and reliability of protein quantification.
Phosphoproteomics analysis builds upon the principles of DIA proteome analysis and incorporates phosphopeptide enrichment strategies. These strategies, such as immobilized metal affinity chromatography (IMAC) or titanium dioxide (TiO2) enrichment, selectively isolate phosphorylated peptides from the complex mixture of proteins. The enriched phosphopeptides are then subjected to LC-MS/MS analysis, enabling the identification and quantification of phosphorylated proteins. Advanced software tools are used to interpret the resulting mass spectra, facilitating the mapping of phosphorylation sites and the determination of phosphorylation stoichiometry.
Technical Advantages of DIA Phosphoproteomics
- Phosphorylation Site Mapping. Studying signaling pathways is made easier by the accurate mapping of phosphorylation sites made possible by DIA phosphorylated proteome analysis.
- Quantification of PTM. Phosphorylation stoichiometry can be measured using this technique, which sheds light on the dynamics of PTMs in various settings.
- Discovery of Biomarkers. DIA phosphorylation proteome analysis helps find disease-specific biomarkers and possible treatment targets by detecting and measuring phosphorylated proteins.
Our Large-Scale Phospho Profiling Services
Our Phosphoproteomics Service offers high sensitivity detection and accurate quantitation of protein phosphorylation and identification of phosphorylation sites. Using Our advanced Discovery Proteomics Platform, we can detect phosphorylation on a large scale from complex biological samples to provide a comprehensive overview of signaling events and pathway activity regulation.
We can analyze up to 9,000 proteins per sample under different conditions, and identify and localize protein phosphorylation sites with or without enrichment approach prior to mass spec analysis. Our Discovery Proteomics Platform is ideal for complete phosphorylation analysis from minimal sample volume. We also offer highly multiplexed targeted proteomics with absolute quantification for customized panels of proteins.
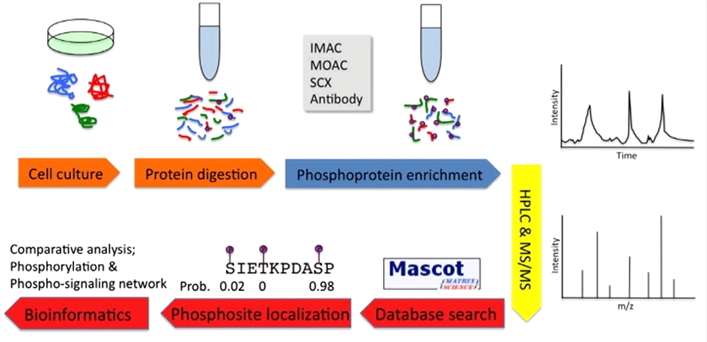 Summary of MS-based phosphoproteomics experiments.
Summary of MS-based phosphoproteomics experiments.
Our DIA Phosphoproteomics Services Include:
- Enzyme Digestion (Cell/Tissue Protein Extraction and Trypsin Digestion, SDS-PAGE and In-Gel Trypsin Digestion).
- Plasma/Serum Protein Depletion.
- Isobaric Labeling.
- Peptide Purification and Concentration.
- Quality Control Runs.
- LC-MS/MS Analysis (DDA Mode).
- Database Search.
Advantages:
- Phospho-protein enrichment (optional).
- Tyrosine, serine and threonine phosphorylation.
- High sensitivity.
- High accurate quantitation of changes in protein phosphorylation between different samples.
- High throughput, more than 9000 proteins can be identified at once.
- High repetition rate.
- Complete and comprehensive data analysis and presentation.
Sample Requirements:
Based on our special protein extraction technology, we can quickly extract proteins from various samples and design personalized experimental schemes according to different experiment purposes. Specific requirements are as follows:
| Sample Type | Protein | # of Cells | Animal Tissue | Plant Tissue | Blood | Urine | Serum | Microbes |
| Quantify | 100 ug | 1×107 cells | 1 g | 200 mg | 1 mL | 2 mL | 0.2-0.5 mL | Dry weighed: 200 mg |
Reference:
- Edmond de Hoffmann.Mass Spectrometry Principles and Applications. 2007.
Case Integrated Phosphoproteomic and Proteomic Analysis Reveals Dysregulated Signaling Networks in Non-Small Cell Lung Cancer.
Background
Non-small cell lung cancer (NSCLC) remains a major global health concern, necessitating a deeper understanding of its molecular mechanisms. Phosphoproteomic and proteomic approaches offer valuable insights into the intricate signaling networks associated with NSCLC, potentially unraveling key biomarkers and therapeutic targets.
Samples
The study incorporated diverse samples, including NSCLC cell lines (PC9, CL68, H3255, CL141, and H1975), MDA-MB-231 breast cancer cell line, and clinical tissues from NSCLC patients. Cell lines were subjected to specific treatments to enhance tyrosine phosphosite abundance. Tissue samples were collected post-surgery from 25 NSCLC patients, ensuring a representative cohort for in-depth analysis.
Technical Methods
Chemicals and Materials:
- Procured chemicals and materials from reputable suppliers, including Sigma Aldrich, Fluka, WAKO, Merck, Promega, 3M, Qiagen, Pierce, Dr. Maisch-GmbH, Synpeptide Co. Ltd, and Biognosys AG.
- Covered a range of reagents, such as formic acid (FA), chloroform, sodium laurate (SL), sodium deoxycholate (SDC), dithiothreitol (DTT), iodoacetamide (IAM), and others essential for various experimental procedures.
Cell Culture and Lysis:
- Utilized human lung adenocarcinoma cell lines (PC9, CL68, H3255, CL141, H1975) and MDA-MB-231 breast cancer cell line.
- Cultured cells in RPMI-1640 medium with specific supplements and treated PC9 and CL68 cells with 250 μM PV (pH 10, with 0.14% H2O2) for 40 minutes to enhance the number of tyrosine phosphosites.
- Collected cells in a lysis buffer cocktail, followed by heat treatment, sonication, and centrifugation for obtaining the lysate.
Lung Cancer Tissue Collection and Lysis:
- Obtained clinical tissues from NSCLC patients with ethical approval and informed consent.
- Processed frozen tissues through rapid thawing, washing with ice-cold PBS buffer, and homogenization in a lysis buffer solution.
- Applied a protein precipitation method with methanol/chloroform and further processed the protein extract for digestion.
Protein Digestion:
- For cell line samples, employed a StageTip-based digestion with Lys-C and trypsin, followed by peptide extraction and fractionation.
- Tissue samples underwent methanol/chloroform protein precipitation, reduction, alkylation, and digestion with Lys-C and trypsin.
- Desalted peptides using reversed-phase StageTips for both cell line and tissue samples.
LC-MS/MS Analysis:
- Employed an LTQ Orbitrap Fusion Lumos Tribrid mass spectrometer coupled with an Ultimate 3000 RSLCnano system.
- Peptides were separated on a Thermo Scientific PepMap C18 column using a gradient of buffer A (0.1% FA in water) and buffer B (0.1% FA in ACN).
- DDA datasets acquired spectra in the Orbitrap with MS resolution of 60,000, and HCD fragmentation for MS/MS.
- DIA datasets utilized a scan range of 400–1250 m/z, MS resolution of 60,000, and HCD fragmentation with specific parameters for MS/MS.
- Applied dynamic exclusion and selected top precursors for MS2 analysis.
Data Processing and Protein Identification:
- Processed DDA data using MaxQuant with Andromeda search engine against the UniProtKB/Swiss-Prot database.
- Constructed hybrid phosphopeptide spectral library from DDA of fractionated cell line and cancer tissue samples using Spectronaut Pulsar search.
- Built an independent proteome spectral library from lung cancer tumor tissues and NSCLC cell lines using MaxQuant and Spectronaut.
- Performed MS2-based quantification, local cross-run normalization, and phosphosite localization using Spectronaut for DIA data.
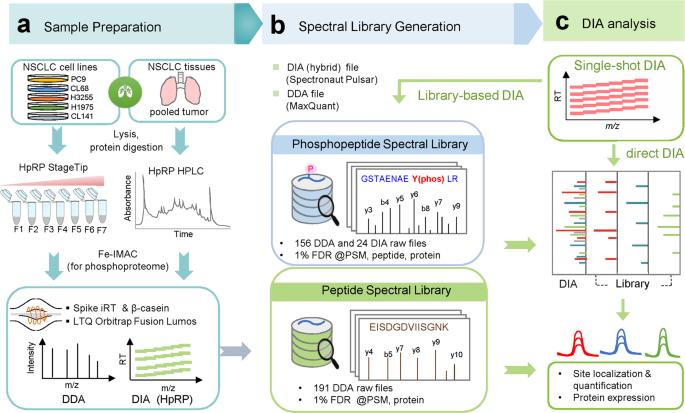 Pipeline for hybrid spectral library construction and phosphosite quantification.
Pipeline for hybrid spectral library construction and phosphosite quantification.
Results
Phosphoproteome Spectral Library Construction: Generated a hybrid library from DDA of fractionated cell line and tissue samples.
Proteome Spectral Library Construction: Constructed a proteome library from lung cancer tumor tissues and NSCLC cell lines.
Differential Phosphoproteomic Analysis: Identified dysregulated phosphosites between NSCLC EGFR-TKI-sensitive and -resistant cell lines.
Tissue vs. Normal Comparison: Detected significant changes in phosphosite abundance between NSCLC tumor tissues and adjacent normal tissues.
Pathway Enrichment Analysis: Performed KEGG pathway analysis, revealing altered signaling pathways in NSCLC.
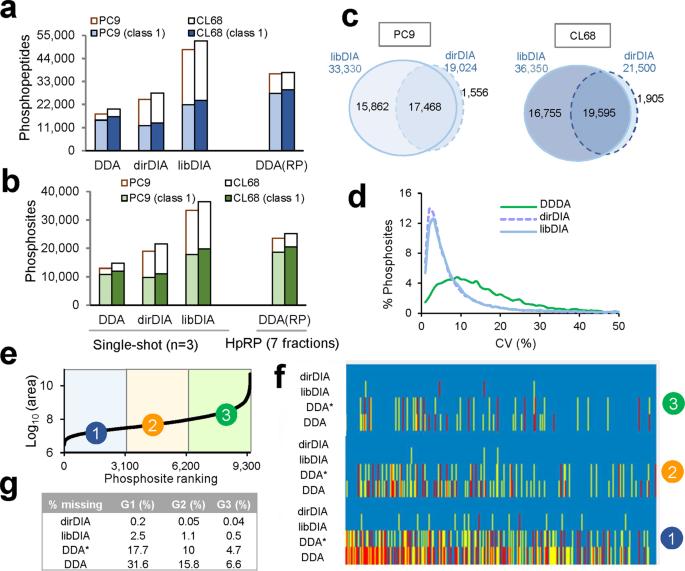 Comparison of quantification performance in DDA and DIA using cell lysate.
Comparison of quantification performance in DDA and DIA using cell lysate.
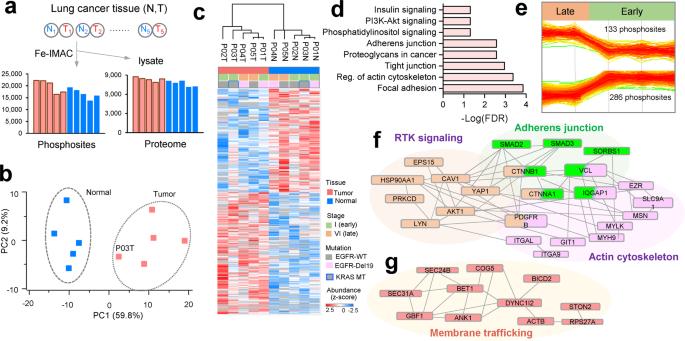 Phosphoproteome profiling of lung cancer tissues.
Phosphoproteome profiling of lung cancer tissues.
Reference:
- Kitata, Reta Birhanu, et al. "A data-independent acquisition-based global phosphoproteomics system enables deep profiling." Nature Communications 12.1 (2021): 2539.

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)