
What is 4D Label-Free Proteomics? The Power of PASEF and Ion Mobility
Traditional 3D Label-Free Quantification (3D-LFQ) relies on three dimensions to identify peptides: Retention Time (RT), Mass-to-Charge ratio (m/z), and Signal Intensity. However, in highly complex biological matrices, multiple peptides often elute at the exact same time with nearly identical masses. These overlapping signals create a high background "noise" that drowns out low-abundance biomarkers, fundamentally limiting the depth of discovery.
As an advanced Ion Mobility 4D Proteomics Service, we introduce a revolutionary fourth dimension: Collision Cross Section (CCS) via Trapped Ion Mobility Spectrometry (TIMS). Before entering the mass analyzer, peptide ions are trapped in a gas flow and physically separated based on their 3D shape and charge (their CCS value). This means that even if two peptides co-elute from the LC column and possess the exact same mass, the TIMS funnel separates them in milliseconds.
When coupled with PASEF (Parallel Accumulation-Serial Fragmentation), the instrument synchronizes the release of these separated ions with ultra-fast quadrupole switching. This dual-funnel technology effectively multiplies sequencing speed and sensitivity by 10x without sacrificing mass resolution. The result is exceptionally clean, high-fidelity MS2 spectra that allow researchers to dig deeper into the "dark proteome" of limited biological samples.
Content Guide
- Principles of 4D Label-Free
- 4D-LFQ vs. 3D-LFQ
- What Problems We Solve
- Service Advantages
- Tailored to Your Needs
- Technology Selection Guide
- Assay Performance
- Workflow
- Sample Requirements
- Bioinformatics & Deliverables
4D-LFQ vs. Traditional 3D-LFQ: Why the 4th Dimension Matters
For B2B research teams, the transition from 3D to 4D proteomics is not just an equipment upgrade; it dictates whether a micro-scale clinical project succeeds or fails due to insufficient data yield.
- Shattering the Input Limit: Traditional 3D instruments typically require 1 to 5 micrograms of purified peptide to achieve a 4,000-protein depth. 4D-LFQ via PASEF can achieve similar or greater depth from as little as 10 to 50 nanograms.
- Cleaning the Matrix: In complex samples like undepleted serum or highly fibrotic tissue, 3D spectra become congested. 4D ion mobility physically isolates the target signals from the chemical noise, resulting in significantly higher unique peptide identification rates.
| Feature | Next-Gen 4D-LFQ (PASEF) | Traditional 3D-LFQ (Standard DDA) |
|---|---|---|
| Minimum Sample Input | Ultra-Low (ng-level, ~1,000 cells) | High (μg-level, ~100,000 cells) |
| Proteome Depth (Low Input) | Extreme (5,000+ proteins easily) | Limited (Often <2,000 proteins) |
| Background Noise Separation | Yes (via Ion Mobility / CCS values) | No (Relies entirely on LC and mass resolution) |
| Sequencing Speed | > 100-120 Hz | ~20-40 Hz |
| Isomer Resolution | High (Separates identical masses by shape) | Poor (Co-eluting isomers form chimeric spectra) |
What Problems Does Low-Input 4D Proteomics Solve?
From rare cell populations to micro-scale biopsies, see how our 4D-LFQ service addresses low-input challenges.

Needle Biopsies & LCM
Capture a comprehensive disease profile from fine-needle aspirates or Laser Capture Microdissection (LCM) tissues rather than just top housekeeping proteins.

FACS-Sorted Cell Populations
Perform deep molecular phenotyping on rare primary immune cells (e.g., <10,000 cells) without the need to pool weeks of sorted samples.

Organoids & 3D Cultures
Utilize PASEF technology to parse individual micro-models, revealing subtle developmental and pharmacological responses with extreme sensitivity.
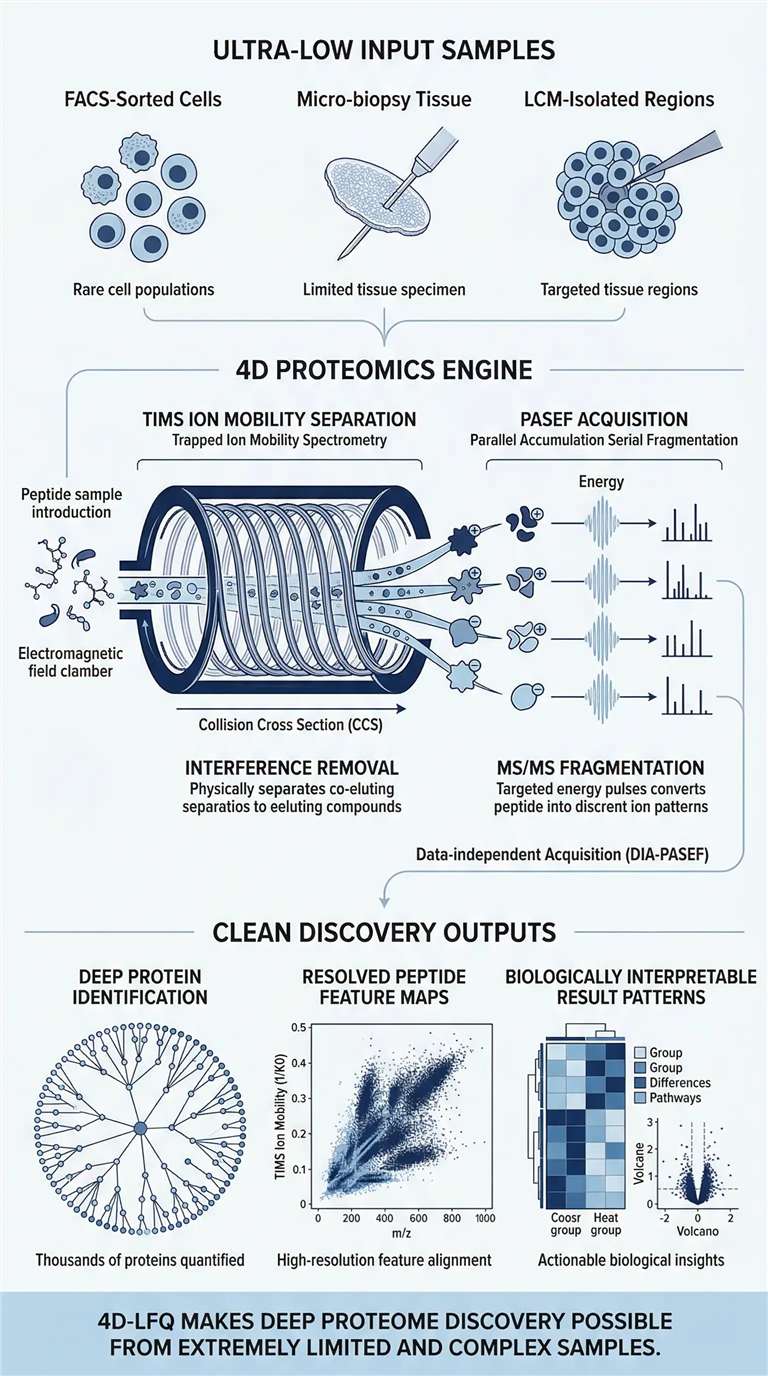
Advantages of Our 4D Label-Free Proteomics Service
4D Ion Mobility (CCS)
Physical gas-phase separation of co-eluting isomeric peptides, dramatically reducing background matrix noise and false discoveries.
PASEF Sensitivity
Breakthrough proteome depth achieved from ng-level biological material that traditional 3D mass spectrometers cannot detect.
Proprietary Low-Input Prep
Optimized micro-scale extraction protocols (e.g., SP3) designed specifically to prevent adsorptive loss in rare cell populations.
Unmatched Reproducibility
Stable quantification across extended sample batches without the ratio-compression artifacts associated with isobaric tagging.
Advanced Bioinformatics
CCS-aware FDR scoring algorithms integrated with deep functional pathway enrichment and biological network analysis.
Seamless Translation
A direct, risk-free pipeline scaling from 4D-LFQ broad discovery directly into targeted 4D-PRM clinical validation panels.
4D Label-Free Proteomics Tailored to Your Needs
Customized extraction, flexible gradients, and targeted bioinformatics.

Micro-Scale Extraction
- Adapted single-pot and microfluidic extraction techniques based on sample type.
- Optimized for highly fibrotic LCM tissues, lipid-rich organoids, or fragile FACS-sorted immune cells to minimize adsorptive losses.

LC Gradient Flexibility
- Extremely short LC gradients (e.g., 15-20 minutes) for high-throughput rapid drug screening.
- Extended multi-hour gradients for ultra-deep profiling of complex lysates and biomarker discovery.
Targeted Bioinformatics
- Customized fold-change cutoffs, imputation methods, and protein-protein interaction (PPI) network analyses.
- Designed to highlight the specific pharmacological or disease pathways relevant to your hypothesis.

Translational Pipeline
- Direct, risk-free scaling from 4D-LFQ broad discovery directly into targeted verification.
- Seamless transition to 4D-PRM clinical validation panels utilizing the same timsTOF architecture.
Technology Selection Guide: 4D-LFQ vs. 4D-DIA vs. TMT
Selecting the optimal proteomics workflow depends heavily on your current project phase, sample volume, and cohort scale. Here is how to navigate the analytical choices:
| Dimension | 4D-LFQ (PASEF DDA) | 4D-DIA (PASEF DIA) | TMT (Isobaric Tagging) |
|---|---|---|---|
| Primary Objective | Deep discovery from ultra-low inputs | Missing value elimination in large cohorts | Highly controlled batch comparison |
| Sample Input Requirement | Extreme micro-scale (ng-level) | Standard (μg-level) | High (requires enough for labeling) |
| Cohort Scalability | Excellent | Infinite | Limited by multiplex channels (e.g., 18-plex) |
| Ideal Project Phase | Early-stage rare cell/biopsy profiling | Large-scale clinical validation | Specific multiplexed drug perturbation studies |
- Choose 4D-LFQ when you are analyzing rare FACS-sorted cells, LCM biopsies, or approaching single-cell equivalents. It excels at mining the deepest possible proteome from minimal starting material where robust quantitative continuity is secondary to maximum identification depth.
- Choose 4D-DIA when scaling up to larger clinical cohorts (e.g., n > 50). It utilizes sequential isolation windows coupled with ion mobility to ensure continuous data acquisition, making it the premier choice for maintaining statistical power across hundreds of samples.
- Choose TMT when comparing 10 to 18 samples in a single batch (e.g., time-course drug treatments) where avoiding run-to-run LC variance and achieving absolute same-batch quantification precision is paramount.
Assay Performance & Validation Specifications
We hold our 4D-LFQ analytical workflows to stringent quality control standards, ensuring your high-throughput data is unassailable and ready for publication:
- Deepest Project-Specific Coverage: Offline 2D-LC fractionated DDA library generation tailored to your exact cohort significantly outperforms generic pan-human libraries by accounting for sample-specific splice variants and background matrix.
- Cohort-Grade Consistency: Implementation of indexed Retention Time (iRT) standards ensures precise chromatogram alignment, maintaining median CVs < 15% across massive multi-month cohort acquisitions.
- Rigorous FDR Control: Advanced statistical algorithms (such as mProphet) are employed for peak scoring and validation, ensuring a false discovery rate (FDR) strictly below 1% at both the peptide and protein levels.
- Expert Support: End-to-end scientific guidance from initial power analysis and cohort study design through to deep biological pathway interpretation.
End-to-End 4D-LFQ Workflow: From Micro-Samples to Deep Insights
Processing nanograms of protein requires more than just high-end hardware; it requires meticulously optimized, ultra-low-loss sample preparation and advanced downstream data parsing. Our comprehensive timsTOF PASEF Proteomics Service follows a rigorous scientific workflow:
Single-pot, solid-phase-enhanced (e.g., SP3), or specialized microfluidic extraction protocols to lyse cells and digest proteins while minimizing surface-adsorption losses.
Nano-flow LC separation coupled with timsTOF PASEF synchronizes MS/MS selection with TIMS elution, generating >100 high-quality spectra per second.
Search engines integrate novel Collision Cross Section (CCS) values alongside mass and fragmentation data, drastically reducing FDR and resolving chimeric spectra.
Normalized matrices undergo PCA, differential volcano plotting, and functional network mapping to highlight actionable biological biomarkers.
Ultra-Low Input Sample Requirements
Our protocols are customized to handle inputs that traditional proteomics facilities routinely reject.
| Sample Type | Standard Input (3D) | Ultra-Low Input (4D Capability) | Preservation State |
|---|---|---|---|
| FACS / Rare Cells | 100,000+ cells | 1,000 - 10,000 cells | Washed cell pellet, flash-frozen |
| Fresh / Frozen Biopsies | 2 - 5 mg | < 0.5 mg (Needle cores) | Flash-frozen in Liquid N2 |
| LCM / FFPE Tissues | 10 curls (10μm) | 1 - 2 curls or targeted LCM areas | Mounted on slides or in tubes |
| Exosomes / EVs | 10 - 20 μg protein | 1 - 5 μg protein | Purified, frozen at -80°C |
Actionable Insights: 4D-Specific Bioinformatics Deliverables
We provide specialized bioinformatics analysis designed to highlight the depth and quality achieved by 4D PASEF technology.
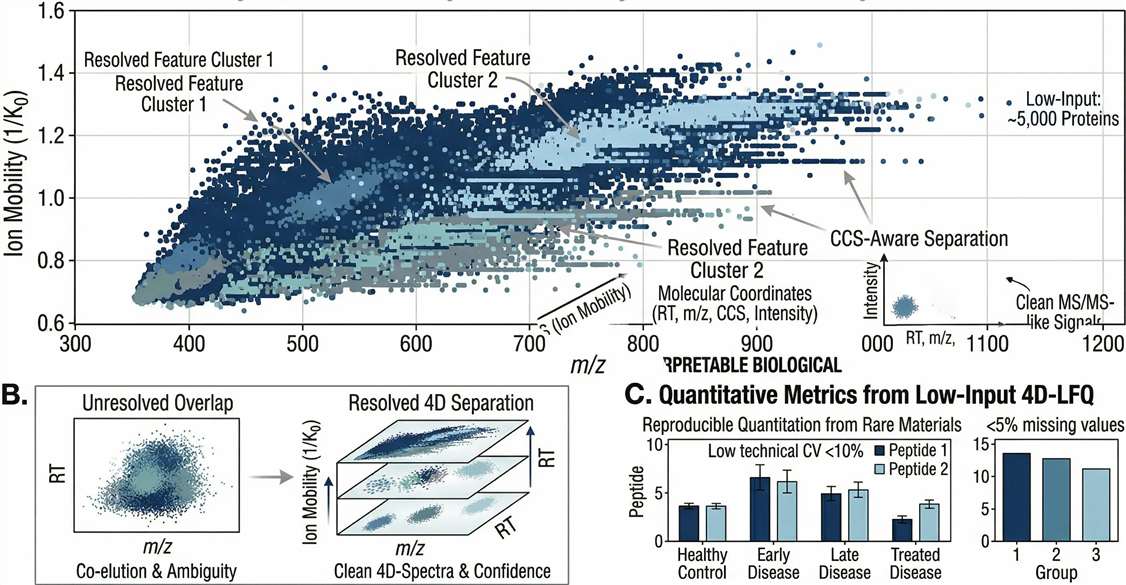
4D Data Quality (CCS Heatmap): m/z versus Ion Mobility (1/K0) heatmaps visually proving the successful physical separation of co-eluting isomeric peptides, guaranteeing high-confidence identifications.
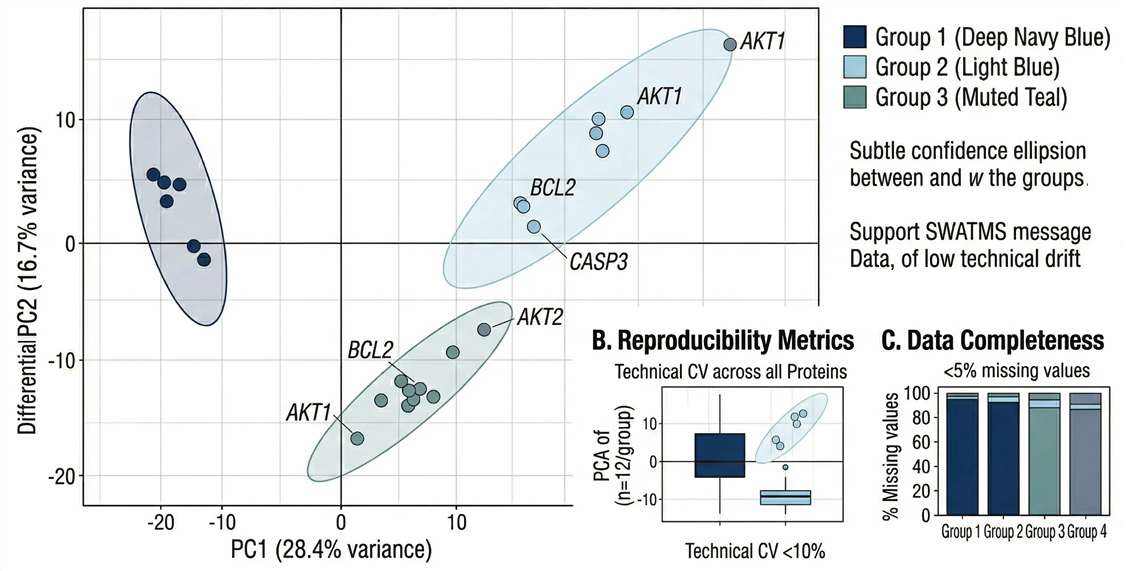
Data Robustness: PCA and CV density plots demonstrating that even with ultra-low inputs, quantitative reproducibility remains tightly controlled.
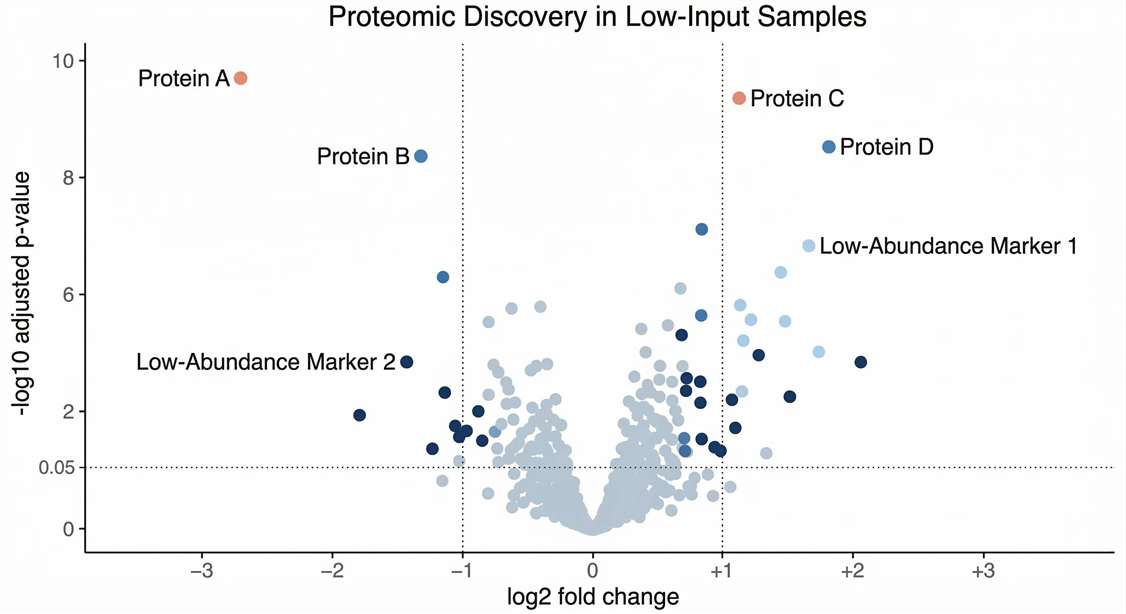
Differential Expression: Volcano plots highlighting the discovery of significantly dysregulated low-abundance proteins that are typically invisible to older mass spectrometers.
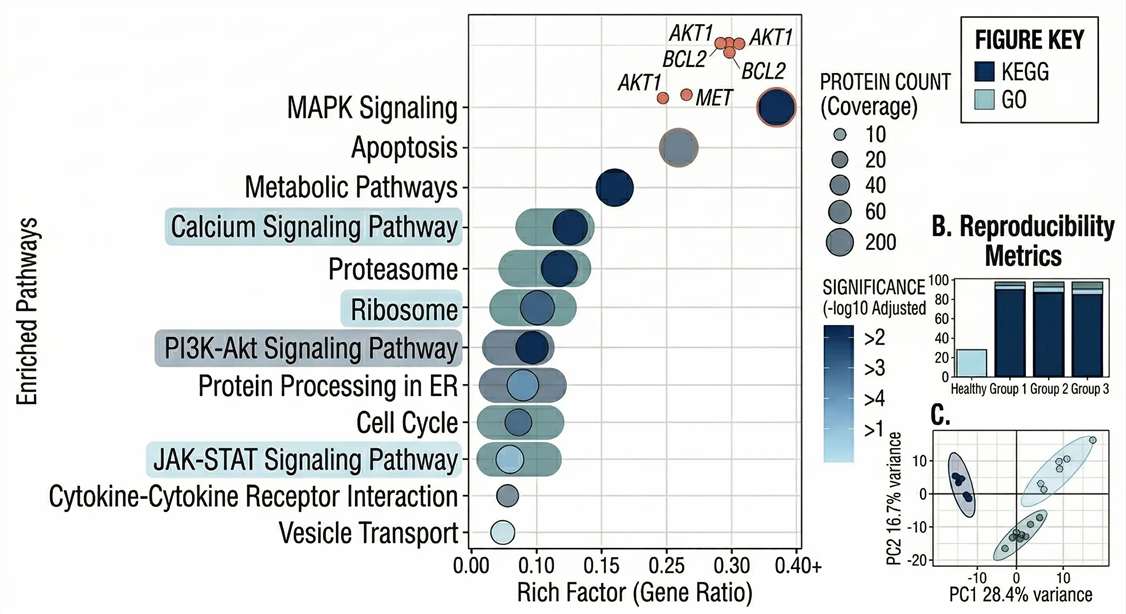
Functional Enrichment: KEGG and GO pathway enrichment mapping to translate raw protein lists into actionable biological mechanisms.
Raw Instrument Files
Native Bruker format files (.d) for independent archival and secondary verification.
4D Filtered Quantitative Tables
High-confidence expression matrices strictly utilizing CCS-aware FDR scoring algorithms.
Comprehensive QC Report
Detailed documentation of extraction efficiency, ID rates, and PASEF cycle parameters.
Publication-Ready Figures
High-resolution graphs and statistical interpretations ready for integration into your next high-impact manuscript.
Deep 4D Proteomics from Challenging FFPE Clinical Specimens
Quantitative proteomics of formalin-fixed, paraffin-embedded cardiac specimens uncovers protein signatures of specialized regions and patient groups
Journal: Nature Cardiovascular Research · Published: 2025
Study Scope
Archived clinical pathology samples are among the most valuable yet technically challenging materials in translational proteomics. Formalin-fixed, paraffin-embedded (FFPE) tissues often suffer from cross-linking, low peptide recovery, and restricted sample input, which historically limited deep proteome analysis.
In this study, researchers demonstrated that high-resolution 4D timsTOF-based proteomics can successfully profile FFPE human cardiac specimens and recover biologically meaningful proteomic signatures across both anatomical regions and patient groups.
- FFPE cardiac tissues were processed for quantitative proteomic analysis and benchmarked against fresh-frozen material.
- The workflow was designed to test whether highly challenging clinical archive samples could still generate deep and reproducible proteome profiles.
- The study further evaluated whether protein-level data from FFPE samples could distinguish both disease status and specialized cardiac regions.
Quantitative Performance and Biological Findings
The study showed that FFPE cardiac specimens remained compatible with deep quantitative proteomics despite their challenging preservation chemistry.
- Approximately 4,000 proteins per sample were quantified from FFPE cardiac tissue.
- Proteomic profiles clearly distinguished disease states and subanatomical regions, demonstrating that archived pathology material can still support high-content biological interpretation.
- The dataset revealed distinct protein signatures associated with specialized cardiac regions and patient-specific pathological variation.
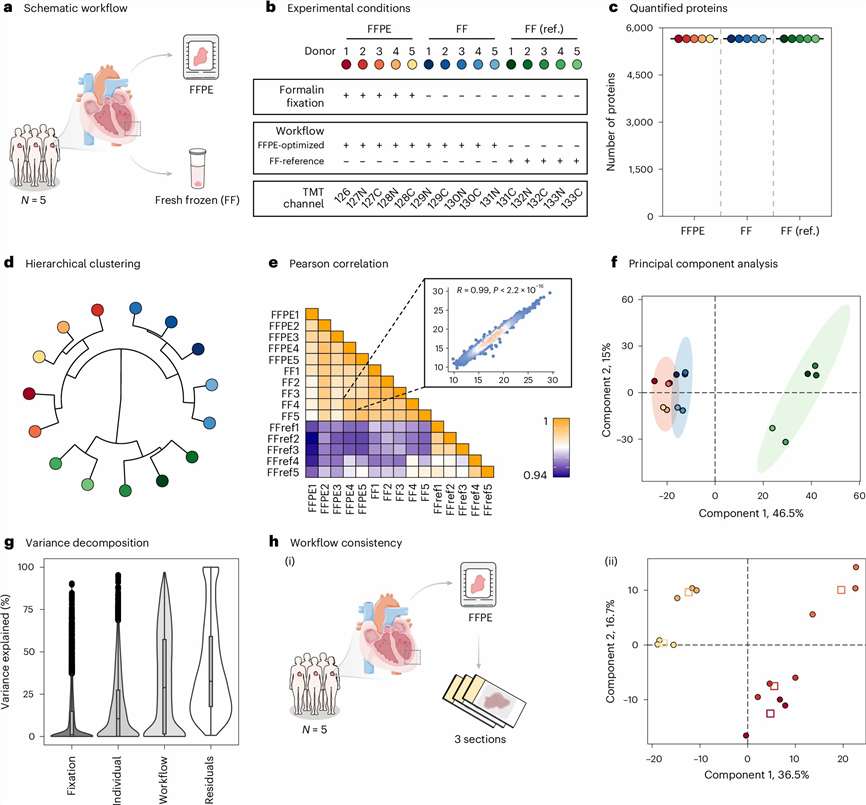 4D timsTOF-based proteomics distinguished low-input FFPE cardiac specimens by both anatomical region and patient group, demonstrating deep and interpretable proteome coverage from archival material.
4D timsTOF-based proteomics distinguished low-input FFPE cardiac specimens by both anatomical region and patient group, demonstrating deep and interpretable proteome coverage from archival material.
Why This Case Matters for Your Strategy
This study is strong supporting evidence for clients considering 4D proteomics on difficult, low-yield, or archived clinical specimens.
It shows that:
- highly challenging FFPE samples can still yield deep proteomic coverage,
- 4D timsTOF-based workflows can recover biologically informative protein signatures from restricted material,
- archived pathology collections can become usable resources for translational discovery rather than unusable legacy assets.
For researchers working with limited clinical biopsies, micro-dissected pathology samples, or precious retrospective archives, this case supports the strategic value of moving to a 4D proteomics workflow when conventional approaches lack depth or sensitivity.
Reference
Achter, J. S., Jensen, T. H. L., Pisano, P., Bundgaard, J. S., Raaschou-Oddershede, D., Rossing, K., Wierer, M., & Lundby, A. "Quantitative proteomics of formalin-fixed, paraffin-embedded cardiac specimens uncovers protein signatures of specialized regions and patient groups." Nature Cardiovascular Research (2025).