Library-Free vs Library-Based DIA Proteomics: Strategies, Software, and Best Use Cases
Data-Independent Acquisition (DIA) has revolutionized proteomics by enabling consistent, deep, and reproducible protein quantification across diverse sample types and study designs. But as the technology evolves, so do the choices researchers must make—chief among them: should you use a library-based DIA workflow, or opt for a library-free approach?
This article provides a detailed comparison of library-based and library-free DIA proteomics strategies—including software options, project suitability, and data delivery practices—designed to help researchers choose the optimal workflow for quantitative proteomics studies.
What is Library-Based DIA?
Library-based DIA relies on the use of empirically derived spectral libraries—comprehensive catalogs of peptide fragmentation patterns generated through high-quality Data-Dependent Acquisition (DDA) experiments. These libraries serve as reference maps against which complex DIA fragment spectra are compared, enabling peptide identification with high specificity and statistical confidence.
How it works: Peptide ions in DIA are fragmented in parallel and matched to the library based on precursor m/z, retention time, and fragment ion intensity patterns using scoring algorithms such as mProphet or Pulsar.
What Goes Into A Spectral Library?
A well-designed spectral library captures:
- Peptide precursor and fragment ion profiles under optimized LC-MS conditions
- Retention time calibration using internal iRT standards
- Instrument-specific fragmentation behavior, ensuring accurate spectral matching
- Biological context, such as tissue-specific expression and common PTMs
At Creative Proteomics, we build project-specific libraries using standardized DDA acquisition schemes optimized for coverage, reproducibility, and spectral clarity. Libraries are generated with rigorous QC protocols, and can be tailored to species, tissue type, experimental conditions, or targeted protein families.
Key Advantages
✓ High identification confidence: Because fragment ion spectra are empirically acquired, the identification of peptides in DIA is based on direct pattern recognition rather than computational inference.
✓ Targeted quantification readiness: Libraries can be enriched for specific proteins, PTMs, or biological pathways, making this approach well-suited for mechanism-of-action studies or panel validation.
✓ Optimal for low-complexity or focused experiments: When the proteome is well-characterized, and sensitivity is critical, library-based DIA maximizes detection efficiency.
Considerations & Limitations
- Sample demand and DDA cost: Generating a high-quality library often requires dedicated samples, which may not be available when working with limited biopsies or irreplaceable clinical materials.
- Biological variability matters: Libraries built from one sample type may not translate well to others. For instance, a library generated from liver tissue may miss key peptides present in tumor or brain samples—even from the same species.
- Experimental inflexibility: Once the spectral library is built, changing sample types, digestion protocols, or gradient lengths may necessitate building a new library from scratch.
Understanding Library Sources: Public, Project-Specific, or Hybrid?
Not all spectral libraries are created equal. Depending on project scope and resource availability, clients may choose between:
- Public spectral libraries (e.g., SWATHAtlas) for common species or tissues—cost-efficient but limited in specificity
- Project-specific libraries, built from matched DDA data—offering maximal depth and biological relevance
- Hybrid strategies, combining public libraries with project-derived DDA for optimal coverage
At Creative Proteomics, we help clients assess sample type, organism, and expected targets to determine the best-fitting library source—balancing coverage, cost, and timeline.
What is Library-Free DIA?
Library-free DIA, also known as directDIA or library-independent DIA, eliminates the need for separate DDA runs by identifying peptides directly from DIA data using computational inference. This approach relies on sophisticated algorithms—often incorporating deep learning models—to predict peptide fragmentation patterns, retention times, and detectability scores based on the amino acid sequence alone.
How it works: Raw DIA data is processed by algorithms that simulate spectra from sequence databases, match observed ion traces to predicted profiles, and score identifications based on statistical models.
Unlike library-based DIA, which depends on previously acquired spectral fingerprints, library-free workflows utilize in silico spectral prediction engines, such as:
- DIA-NN: Combines deep neural networks with fast spectral deconvolution, allowing high-throughput analysis with high identification rates.
- MSFragger-DIA + FragPipe: Uses open search to discover unexpected PTMs and tolerates wide precursor windows.
- Prosit-enabled workflows: Leverage trained neural networks to generate highly accurate fragment ion intensity patterns and retention time predictions.
Key Advantages
✓ No need for prior DDA: Particularly beneficial when sample quantity is limited or high-throughput speed is essential.
✓ Highly scalable: Library-free DIA excels in large-scale studies involving hundreds of samples, such as time-course experiments or patient cohorts.
✓ Compatible with novel proteomes: When working with non-model organisms, microbiomes, or edited proteomes, library-free approaches bypass the need for matched reference spectra.
✓ Flexible pipeline design: Allows for real-time sample addition, variable gradient lengths, or experimental conditions without regenerating a new library.
Considerations & Limitations
- Algorithm-dependent performance: The quality of results hinges on the software's ability to predict spectral features with high fidelity. Misalignment in retention time or inaccurate fragmentation predictions may affect identification rates.
- Greater need for QC interpretation: Since identifications are computationally inferred, clients should evaluate quality metrics (e.g., FDR, match score distributions, peak picking reproducibility) more critically.
- Lower specificity for low-abundance targets: In highly complex samples, library-free workflows may miss borderline signals that would otherwise be recovered through spectral matching.
Creative Proteomics Insight: For projects where biological complexity is high and discovery breadth matters more than validation specificity, we recommend library-free DIA as the default strategy—unless pre-existing DDA data justifies otherwise.
👉 Learn more about our DIA Quantitative Proteomics Service and how we tailor workflows for large-scale discovery and reproducible quantification.
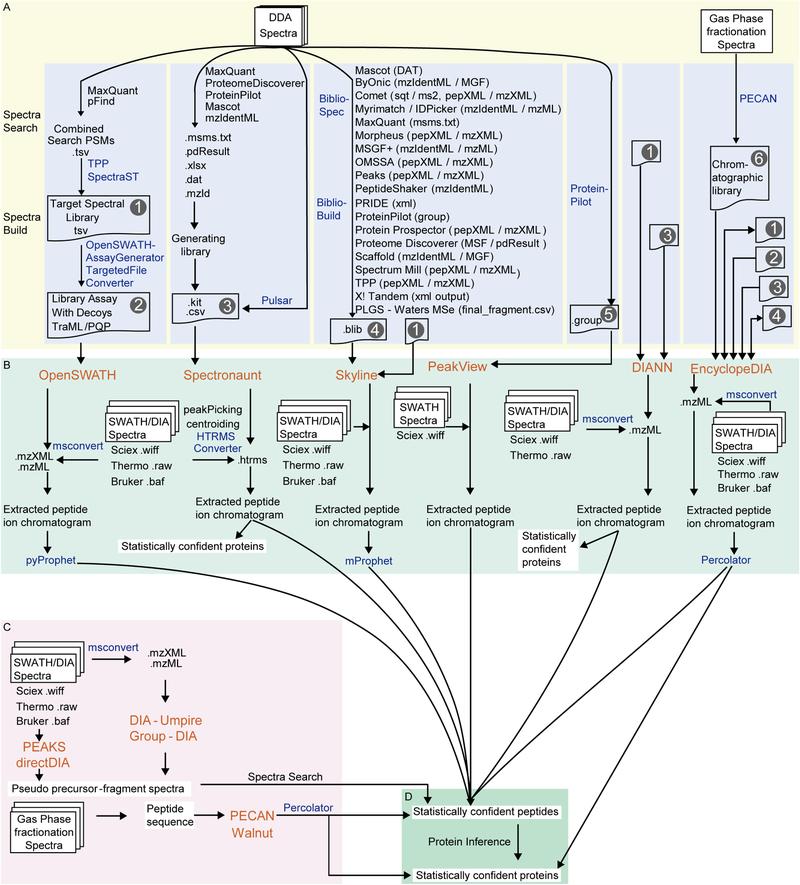 Comparison of DIA data analysis workflows. Panels A–B show library-based approaches using DDA-derived spectral libraries with tools like Spectronaut, Skyline, and OpenSWATH. Panel C depicts library-free methods such as directDIA and DIA-Umpire, which extract peptide information directly from DIA data. Panel D highlights protein inference tools applied after peptide identification (Zhang, Fangfei, et al., 2020).
Comparison of DIA data analysis workflows. Panels A–B show library-based approaches using DDA-derived spectral libraries with tools like Spectronaut, Skyline, and OpenSWATH. Panel C depicts library-free methods such as directDIA and DIA-Umpire, which extract peptide information directly from DIA data. Panel D highlights protein inference tools applied after peptide identification (Zhang, Fangfei, et al., 2020).
Library-Based vs Library-Free DIA: Strategic Comparison
| Feature | Library-Based DIA | Library-Free DIA |
| Prior DDA Requirement | ✓ Yes | ✕ No |
| Spectral Library Source | Project-specific or public | Inferred via algorithm |
| Initial Setup Time | Longer (DDA + QC) | Shorter |
| Sample Demand | Higher (≥2 runs/sample) | Lower |
| Ideal Project Type | Targeted validation, pathway-focused | Discovery, large-scale profiling |
| Organism Compatibility | Well-characterized species | Broad, including novel or non-model |
| Flexibility (e.g. gradient/method changes) | Limited | High |
| Software Options | Spectronaut, Skyline, Scaffold | DIA-NN, FragPipe, MSFragger |
| Identification Specificity | Very high (empirical match) | High (model prediction, depends on QC) |
| Low-Abundance Protein Detection | Excellent if in library | Moderate unless optimized |
| Panel Customization Support | Strong (preselected targets) | Good (flexible post hoc selection) |
| QC Interpretation Complexity | Lower | Higher (due to inference) |
| Public Dataset Compatibility | High (if library exists) | Medium (requires reprocessing) |
| Turnaround Time | Moderate to long | Fast |
| Best Use Case Example | Known biomarker panel across replicates | Multi-sample discovery in new condition |
When to Use Each Strategy
Library-Based DIA is optimal when:
- You are validating known targets or biomarkers, such as in a pathway-focused study or follow-up to RNA-seq findings
- You have access to high-quality, sufficient sample material, enabling dedicated DDA runs
- You require precise quantification of specific protein groups, such as kinase families, transcription factors, or PTM-modified peptides
- Your regulatory or collaboration framework requires fixed methods, as in some biotech and pharma partnerships
Example: Quantifying a predefined panel of neuroinflammatory markers across treated vs untreated brain samples in a preclinical model.
Library-Free DIA is ideal when:
- You're exploring complex or poorly characterized systems, such as tumor microenvironment, microbiome proteomics, or plant-microbe interactions
- Sample material is limited, e.g., FFPE slices, organoid lysates, or microdissected tissues
- The study includes >20 samples, such as patient cohorts, time-course studies, or drug screening assays
- You need rapid turnaround without additional MS runs, ideal for pilot screens, grant support studies, or early R&D phases
Example: Running discovery proteomics on 40 patient plasma samples to identify early metabolic response signatures.
What You Receive: Tailored Deliverables for Maximum Usability
At Creative Proteomics, we focus not only on generating high-quality DIA data but also on delivering results that are accessible, interpretable, and ready for immediate use—whether for internal decision-making, publication, or bioinformatics integration.
Core Deliverables
| Content | Format | Notes |
| Protein/Peptide Quant Tables | .xlsx, .csv | Abundance data with annotations (UniProt ID, gene name, sequence, CV%) |
| Raw MS Files | .raw, .mzML | Compatible with vendor and open-source tools |
| ID Reports | .tsv, .pdf | PSMs, scores, FDR filtering, RT alignment data |
| QC & Visualization Plots | .pdf, .html | PCA, volcano, heatmaps, replicate correlation, chromatogram overlays |
| Optional Enrichment Analyses | .xlsx, .svg | GO, KEGG, PPI, functional clusters |
Each delivery includes a README and method summary, with clear documentation of software versions, search parameters, and filtering thresholds—ensuring transparency and reproducibility.
Customization & Add-ons
- Format conversions for Prism, R, or GraphPad
- Interactive HTML dashboards (filterable by protein/condition)
- Journal-ready figures with statistical labeling
- Summary slides for internal reviews or grant reporting
- Optional interpretation session with our proteomics specialists
Data Access & Security
- Secure download link with project-specific access
- Version-controlled file packages with unique identifiers
- Long-term archiving options available upon request
References:
- Zhang, Fangfei, et al. "Data‐independent acquisition mass spectrometry‐based proteomics and software tools: a glimpse in 2020." Proteomics 20.17-18 (2020): 1900276.
- Gillet, Ludovic C., et al. "Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis." Molecular & Cellular Proteomics 11.6 (2012): O111-016717.
- Gessulat, Siegfried, et al. "Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning." Nature methods 16.6 (2019): 509-518.
- Bruderer, Roland, et al. "Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results." Molecular & Cellular Proteomics 16.12 (2017): 2296-2309.
- Guo, Tiannan, et al. "Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps." Nature medicine 21.4 (2015): 407-413.

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)