Avoiding Failure in DIA Proteomics: Common Pitfalls and How to Fix Them
Why DIA Projects Fail—And Why It Matters to Fix Them
Data-Independent Acquisition (DIA) has emerged as a powerful tool for comprehensive, high-throughput proteomic profiling. Yet despite its technical strengths—deep coverage, reproducibility, and scalability—DIA is not immune to failure. In fact, when improperly executed, it can produce misleading results that derail entire studies, especially in translational or biomarker research contexts.
Common issues such as inadequate sample preparation, poor spectral library design, and faulty data interpretation can all result in reduced peptide identification, low reproducibility, or biologically implausible quantification trends. These failures aren't always obvious—some are masked until downstream analyses (e.g., differential expression or pathway enrichment) yield contradictory or irreproducible outcomes.
For CROs, pharma partners, or academic labs under pressure to deliver meaningful proteomic insights, avoiding such pitfalls is not merely a technical preference—it's a matter of scientific integrity, budget efficiency, and project viability.
This technical resource breaks down common reasons why DIA experiments fail, how to recognize red flags early, and—most importantly—how Creative Proteomics helps prevent and correct these issues through expert-led QC workflows and transparent reporting.
| Pitfall Type | Typical Consequence | Recoverability |
| Low peptide yield | Reduced ID count, poor quantification | Partial |
| Library mismatch | Missed targets, low specificity | High (rebuild) |
| Acquisition misconfig | Overlapping windows, poor resolution | Medium |
| QC oversight | Inconsistent replicates, high CV% | Low |
Sample-Related Failures: The Root of Downstream Noise
The most common point of failure in a DIA proteomics project begins at the sample level. Unlike DDA workflows, which selectively trigger fragmentation on the most abundant precursors, DIA continuously fragments all ions within predefined m/z windows—capturing a complete picture, but also amplifying any upstream variability. If a sample is poorly extracted, insufficiently digested, or chemically contaminated, no software algorithm can rescue the signal quality. These foundational errors directly compromise peptide detectability, quantification linearity, and statistical power downstream.
Common Pitfalls in Sample Handling
| Issue | Description | Impact |
| Low peptide yield | Under-extraction from FFPE, fibrous tissue, or microdissected samples | Weak total ion current, poor ID rate |
| Incomplete digestion | Denaturation/reduction/alkylation skipped, causing missed cleavages | Lower match confidence, increased FDR |
| Chemical interference | Salts, detergents, or lipids retained post-extraction | Suppressed ionization, poor RT alignment |
Peptide integrity and digestibility are particularly critical in DIA, where incomplete enzymatic cleavage leads to ambiguous fragment assignments and suboptimal quantification. Likewise, impurities such as heme, SDS, or ethanol residues can cause retention time drifts and coelution artifacts—especially detrimental in complex plasma or organoid samples.
How We Address It at Creative Proteomics
To minimize pre-analytical errors, we enforce a three-tier sample qualification checkpoint before DIA runs:
- Protein Concentration Check – Measured via BCA or NanoDrop; low input flags under-extracted matrices.
- Peptide Yield Assessment – Digest yield quantified to ensure sufficient material for MS injection.
- LC-MS Scout Run – Performed on a subset digest to preview peptide complexity, retention time spread, and ion abundance distribution.
These QC steps enable us to flag potential issues before full acquisition, allowing clients to adjust upstream protocols or submit fresh material if needed. For challenging matrices, such as FFPE or bioreactor supernatants, we offer optimized extraction kits and optional preprocessing services.
Tip for Clients
High-risk samples include those from:
- Archival FFPE tissues
- Extremely low-input organoids (<5 μg)
- Blood components without prior albumin depletion
Acquisition Parameter Pitfalls: Suboptimal MS Settings Undermine DIA Quality
Even when sample preparation is flawless, poorly configured mass spectrometry parameters can sabotage the success of a DIA experiment. Unlike DDA, where instrument settings are dynamically adjusted in real-time, DIA acquisition relies on pre-defined scan schemes. If those schemes are mismatched to sample complexity or chromatography conditions, signal overlap, quantitation errors, and identification loss will follow.
Typical Parameter Missteps
| Problem | Description | Consequence |
| SWATH windows too wide | Overly broad m/z ranges per window lead to mixed fragment ions | Poor selectivity, chimeric spectra |
| Inadequate scan speed | MS2 acquisition not fast enough for LC peak width | Missing peptide apexes, reduced quant accuracy |
| Short gradients | Peptides elute too close, complicating separation | Coelution artifacts, poor RT alignment |
| Copy-paste DDA settings | Using DDA-oriented collision energies or resolutions | Suboptimal fragmentation, reduced signal-to-noise |
In particular, wide isolation windows—sometimes applied for speed—can cause excessive precursor interference, especially in plasma or tissue lysates. Likewise, fast gradients (<30 minutes) often compress chromatographic resolution beyond the instrument's capacity to distinguish individual peptides in the cycle time allotted.
Creative Proteomics Solutions
At Creative Proteomics, our acquisition protocols are optimized across multiple platforms (Thermo Exploris, Bruker timsTOF, SCIEX ZenoTOF) and sample classes. Key practices include:
- Adaptive window schemes: Dynamic SWATH window design based on peptide density predictions
- Cycle time calibration: Tailored MS2 scan rates to match LC peak width (~8–10 points per peak)
- Retention time anchoring: Use of indexed retention time (iRT) peptides in all runs
- Platform-specific optimization: DIA with high-mobility separation tuning or Zeno-based DDA-DIA hybrid for improved PTM capture
We also offer a DDA–to–DIA migration consult, helping clients adjust legacy settings from DDA-based workflows (collision energies, resolutions, fill times) to suit the broader coverage demands of DIA.
Client Tip: Checklist for Acquisition Readiness
- Is your LC gradient ≥ 45 min for complex samples?
- Are your SWATH windows < 25 m/z on average?
- Is your cycle time ≤ 3 sec to ensure peak sampling density?
- Are you reusing DDA methods? If yes, ask us for a compatibility review.
Spectral Library Missteps: When Matching Fails You
In library-based DIA workflows, the quality and relevance of the spectral library directly determine the success of peptide identification and quantification. While public or pre-built libraries offer convenience, mismatches in species, tissue type, or instrument conditions can severely degrade performance—leading to low identification rates, inflated false discovery rates, or biologically meaningless results.
Common Library Pitfalls
| Issue | Description | Consequence |
| Tissue-library mismatch | Using a liver-derived spectral library for brain tissue or tumor lysates | Missed key biomarkers, poor coverage |
| Species incompatibility | Applying human libraries to mouse or custom strains | Low match confidence, drop in ID count |
| DDA quality issues | Libraries built from low-resolution or poorly fractionated DDA runs | Incomplete fragment coverage, ambiguous identifications |
| Fixed gradient bias | Libraries created under different LC gradients than your DIA run | RT drift, misalignment in peak integration |
Even minor inconsistencies—such as a gradient shift from 90 to 60 minutes—can distort peptide elution times enough to hinder accurate library matching. Worse yet, project timelines can suffer if new DDA runs are required to rebuild libraries mid-way through a study.
When to Use Project-Specific vs. Public Libraries
| Library Type | Coverage | Biological Relevance | Turnaround Time | Recommended Use |
| Public (e.g., SWATHAtlas) | Moderate | Generic | Fast | Common cell lines, method development |
| Project-specific | High | Matched to sample | Longer | Complex tissues, biomarker discovery |
| Hybrid (public + custom DDA) | High | Balanced | Medium | Semi-exploratory with known targets |
At Creative Proteomics, we help clients evaluate whether their study justifies a dedicated DDA library or would benefit more from a library-free approach using tools like DIA-NN or MSFragger-DIA. This assessment is based on three key variables:
1. Sample complexity (e.g., plasma vs. single-cell lysates)
2. Biological novelty (e.g., model organism vs. human tissues)
3. Project goals (targeted quantification vs. discovery)
Our Library-Building Standards
To ensure high identification confidence, Creative Proteomics constructs DDA-based spectral libraries with:
- ≥2 replicate DDA runs per sample type, under matching LC gradients
- Inclusion of iRT standards for consistent retention time calibration
- Rigorous peptide FDR filtering and protein inference scoring
- Fragment coverage completeness metrics to assess quantifiability
Software and Interpretation Errors: Missteps in Data Analysis
Even with flawless samples and acquisition, DIA proteomics results can fall apart at the analysis stage if the software pipeline is mismatched, misconfigured, or misinterpreted. While DIA's strength lies in comprehensive data capture, its complexity demands informed tool selection and statistically sound parameter setting. Unfortunately, this is where many projects falter—often quietly.
Common Software-Related Pitfalls
| Issue | Description | Typical Impact |
| Inappropriate software selection | Using software unsuited for your library type (e.g., library-based tools on library-free datasets) | Incomplete identifications, inflated FDR |
| Misconfigured parameters | Default FDR thresholds, missed decoy calibration, improper RT alignment settings | False positives, peak misassignment |
| Poor understanding of output | Misreading volcano plots, over-reliance on fold-change alone | Misleading biological interpretation |
Without experienced guidance, these errors often go undetected until they impact downstream analysis—such as inconsistent pathway enrichment or unexpected replicate clustering.
Tool Selection Should Match Experimental Design
| Project Feature | Recommended Tool(s) |
| Library-free DIA | DIA-NN, MSFragger-DIA, FragPipe |
| Project-specific spectral library | Spectronaut, Skyline, Scaffold DIA |
| Need for open search or PTM profiling | MSFragger-DIA, PEAKS, EncyclopeDIA |
| Emphasis on statistical control and transparency | Scaffold DIA, Spectronaut (report builder) |
At Creative Proteomics, we pre-screen each project's scope and sample type to determine the optimal software pipeline. Our standard workflows are designed to balance:
- Speed for high-throughput projects
- Depth for exploratory studies
- Traceability for regulated or multi-party projects
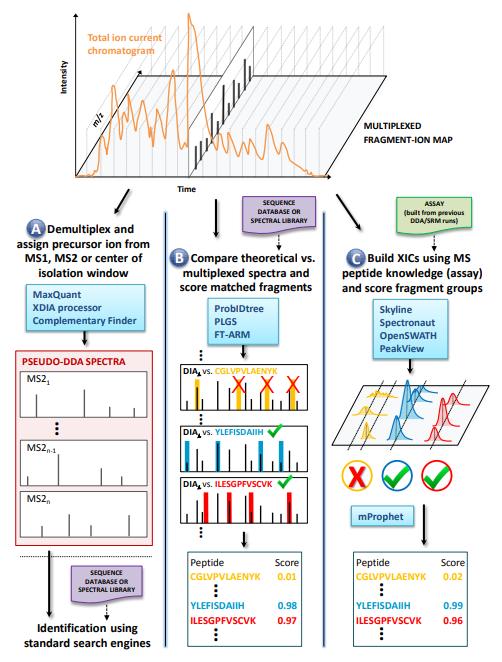 Figure 2. Overview of DIA Data Processing Workflows (Bilbao, Aivett, et al., 2015).
Figure 2. Overview of DIA Data Processing Workflows (Bilbao, Aivett, et al., 2015).
A) Pseudo-DDA generation via demultiplexing; B) Direct matching of multiplexed spectra using databases or libraries; C) Targeted XIC extraction using prior spectral libraries. Software tools are shown in blue for each strategy.
Fixing Common Issues: A Phase-by-Phase Strategy
While DIA proteomics offers powerful breadth and consistency, its success hinges on vigilance across every step—from sample prep to final report. Rather than relying on post-hoc troubleshooting alone, Creative Proteomics integrates preventive strategies at three critical phases: sample preparation, acquisition, and data analysis.
Phase 1: Sample Preparation Safeguards
| Control Point | Strategy | Benefit |
| Protein quantification | BCA or NanoDrop validation with predefined thresholds | Avoids underloading and ion suppression |
| Digest QC | LC-MS scout run of test digest to assess missed cleavages, signal distribution | Prevents poor peptide representation |
| Contaminant screening | Checklists for detergent residues, blood contamination, salts | Ensures ionization consistency |
Internal Standard Use: We recommend internal iRT peptides in every digest to monitor LC consistency and retention time alignment from the earliest stage.
Phase 2: DIA Acquisition Optimization
Our lab implements instrument-specific, project-optimized DIA acquisition templates, built from extensive benchmarking. This includes:
- SWATH window calibration for each LC gradient length and column type
- MS2 injection time balancing for high-complexity samples
- Dynamic exclusion filters to reduce redundant fragmentation of dominant ions
Before batch runs, test injections are performed, with metrics like Total Ion Current (TIC) uniformity, precursor coverage, and signal-to-noise ratios reviewed by senior MS analysts.
💡 Need a platform recommendation? We guide clients on choosing between Orbitrap, TOF, or ion mobility-enhanced systems based on study goals—not just instrument availability.
Phase 3: Data Processing & Validation
| Step | Creative QC Action |
| Protein identification | Use of multiple FDR thresholds (1%, 0.1%) for layered review |
| Quantification | Coefficient of Variation (CV) filtering < 20% across replicates |
| Normalization | Intensity-based or iRT-based normalization depending on sample type |
| Batch assessment | PCA and replicate clustering included by default |
| Final review | Manual inspection of outliers, volcano plots, ID depth, and RT shifts |
All pipelines undergo cross-platform benchmarking (e.g., comparing DIA-NN and Spectronaut outputs) for projects involving novel organisms, low-input samples, or challenging PTM enrichment.
Creative Proteomics QA/QC Workflow: Built-in Risk Prevention
Ensuring reproducible, high-confidence DIA proteomics data requires more than instrumentation and software—it demands a structured, quality-centered workflow. At Creative Proteomics, we integrate multi-point QA/QC checkpoints throughout the entire project cycle, minimizing failure risks from sample intake to data delivery.
Quality Assurance Across Key Stages
| Stage | Quality Measures | Purpose |
| Sample Intake | Protein quantification (e.g., BCA), peptide yield estimation, digest quality check | Ensures input meets minimum quality standards for downstream DIA |
| Instrument Calibration | Retention time monitoring, TIC stability, MS/MS signal inspection via standard runs | Confirms LC-MS/MS performance and run-to-run reproducibility |
| Library Validation (if applicable) | Library-sample match verification, decoy/target evaluation | Verifies that the spectral library suits the sample type and study objective |
| Data QC & Filtering | Peptide/protein ID count, CV%, FDR assessment, PCA clustering | Confirms biological and technical reproducibility |
| Report Review & Delivery | Internal technical review, checklist-based reporting | Delivers fully annotated, quality-verified results with clear documentation |
Note: For library-free workflows, library validation is replaced by in-silico model quality monitoring, including predicted RT alignment and peptide detectability scoring.
What We Deliver (With Quality Metrics Included)
- Raw MS files (.raw, .mzML)
- Quantification tables (protein and peptide level, with CV%, annotations)
- ID summary reports (FDR thresholds, decoy validation, matched fragment statistics)
- QC visualization suite:
- PCA plots
- Heatmaps and volcano plots
- Protein count per sample and replicate correlation matrix
Each dataset includes a README document outlining methods, QC checkpoints, and software versioning—for full transparency and easier publication or secondary analysis.
Handling QC Issues
In cases where data do not meet expected QC benchmarks (e.g., low ID count, poor replicate correlation), we proactively flag these issues and consult the client before proceeding. Depending on root cause and remaining sample availability, re-analysis or re-acquisition may be recommended and executed after client confirmation.
We do not promise automatic re-runs, but offer scientifically justified solutions for data improvement within project constraints. Our goal is not just data delivery—but data clients can trust.
References:
- Bilbao, Aivett, et al. "Processing strategies and software solutions for data‐independent acquisition in mass spectrometry." Proteomics 15.5-6 (2015): 964-980.
- Barkovits, Katalin, et al. "Reproducibility, specificity and accuracy of relative quantification using spectral library-based data-independent acquisition." Molecular & Cellular Proteomics 19.1 (2020): 181-197.
- Doellinger, Joerg, et al. "Isolation window optimization of data-independent acquisition using predicted libraries for deep and accurate proteome profiling." Analytical chemistry 92.18 (2020): 12185-12192.
- Wen, Bo, et al. "Carafe enables high quality in silico spectral library generation for data-independent acquisition proteomics." bioRxiv (2024).
- Yu, Fengchao, et al. "Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform." Nature communications 14.1 (2023): 4154.
- Brenes, Alejandro J. "Calculating and Reporting Coefficients of Variation for DIA-Based Proteomics." Journal of Proteome Research 23.12 (2024): 5274-5278.

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)