
Key takeaways
- In LC–MS/MS proteomics, apparent EV yield is often a trap—background proteins and lipoproteins can dominate ion current and bury low-abundance EV biomarkers.
- The most MS-reliable exosome isolation for LC–MS proteomics workflows are built around contaminant separation (SEC polishing; orthogonal density steps) and process controls (pre-analytics, fraction mapping, QC gates).
- Sample matrix dictates failure modes: plasma/serum demands lipoprotein-aware strategies; urine requires uromodulin control; conditioned media requires scalability plus depletion of serum-derived particles.
Why High-Purity Isolation is the Bottleneck in Exosome Isolation for LC–MS Proteomics
In extracellular vesicle (EV) proteomics, the hardest part isn't getting "particles"—it's getting a preparation that lets your LC–MS/MS see what you actually care about.
Blood, urine, and conditioned media contain abundant non-EV material: soluble proteins, protein complexes, and (in plasma) massive numbers of lipoproteins. In the mass spectrometer, these high-abundance species can dominate the ion population and suppress signals from low-abundance EV membrane proteins and tissue-derived biomarkers. The result is a familiar pattern: your nanoparticle tracking analysis (NTA) looks great, but peptide IDs drop, quantitative CVs inflate, and the biology gets noisy.
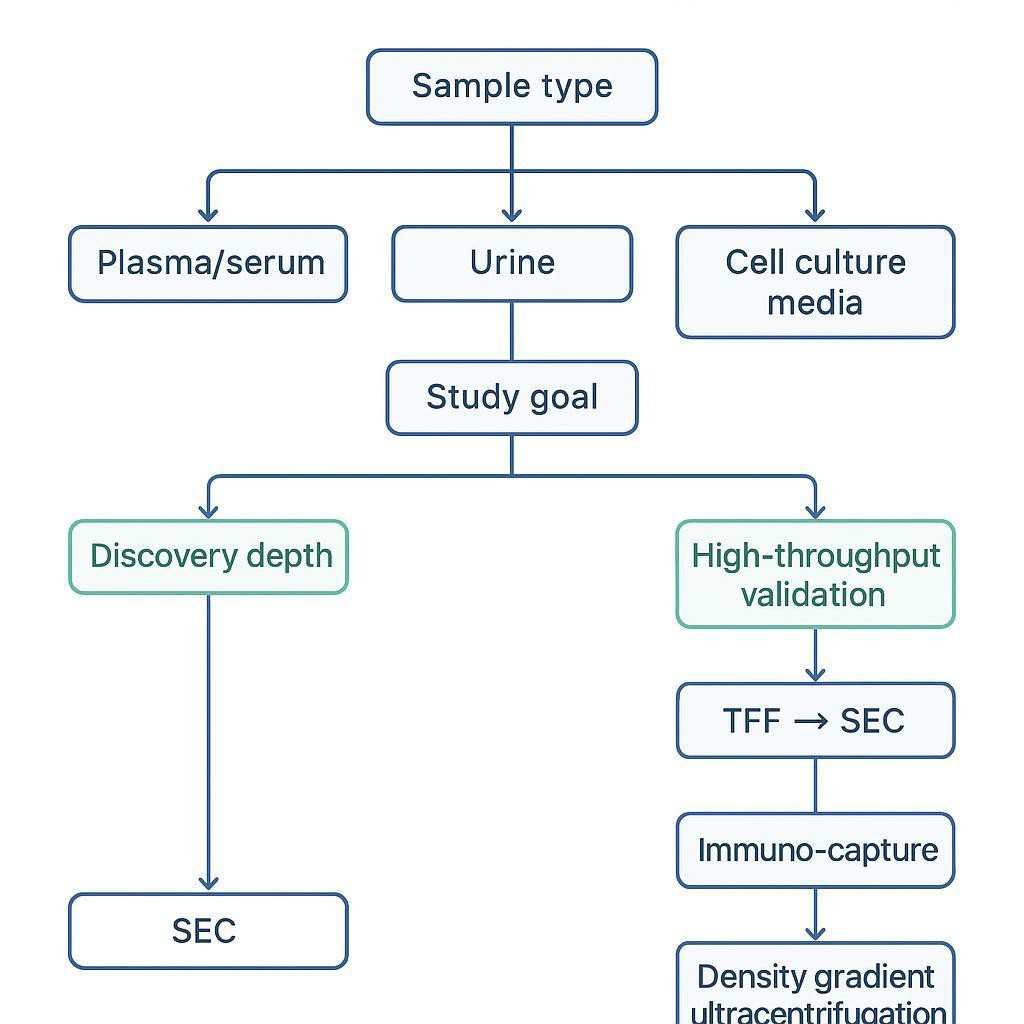
A practical way to think about EV isolation for proteomics is this:
Key Takeaway: If your isolation method optimizes particle count without controlling co-isolated protein/lipoprotein background, it can reduce LC–MS depth—even when the "yield" metric improves.
Reviews comparing isolation strategies consistently highlight size-exclusion chromatography (SEC) as a purity-forward option for downstream omics, especially when paired with upstream concentration that doesn't over-stress vesicles or concentrate contaminants indiscriminately (e.g., tangential flow filtration).
If you're building a pipeline specifically for exosome isolation for LC–MS proteomics, it's useful to treat each unit operation as serving one of two goals: (1) concentrate vesicles without deforming them, and (2) actively remove the contaminants that otherwise dominate MS signal.
The rest of this article is an engineering-minded roadmap: what breaks in each sample type, what workflows tend to recover MS-ready EVs, and how to decide between discovery-depth methods and higher-throughput validation paths.
Plasma and Serum: Decoupling EVs from Lipoproteins
Plasma and serum are the most demanding matrices for EV proteomics because EVs are surrounded by a dense "sea" of lipoproteins and protein complexes. The core analytical problem is overlap: lipoproteins can be similar to EVs in size and buoyant density, so many common workflows co-isolate them.
This is not a cosmetic contamination issue. Lipoprotein-associated apolipoproteins and other abundant plasma proteins can overwhelm LC–MS/MS acquisition and complicate interpretation of "EV signatures."
Pre-Analytics and Pre-Clear Steps
Before you touch any high-speed step, you can win (or lose) a large fraction of your downstream MS quality.
1) Govern the sample class: plasma vs serum
- Plasma retains clotting factors and can be more stable for certain workflows, but platelet-derived particles can be a major confounder if pre-clearing is weak.
- Serum can include vesicles released during coagulation; if you're using serum, a standardized clotting procedure—and controlling clot-related carryover—matters for comparability.
2) Remove platelets and cell debris deliberately For LC–MS proteomics, platelet carryover is a classic failure mode: it inflates particle counts and biases the proteome toward platelet-related proteins.
Operationally, pre-clear is less about "one magic g-force" and more about consistency:
- Use a staged low-speed centrifugation approach to remove cells/debris and reduce platelets.
- Avoid harsh mixing and repeated freeze–thaw cycles that can generate artifacts.
3) Treat "yield" metrics as conditional If your early steps leave platelets/lipoproteins unchecked, the later "high-yield" isolation may simply concentrate the wrong things.
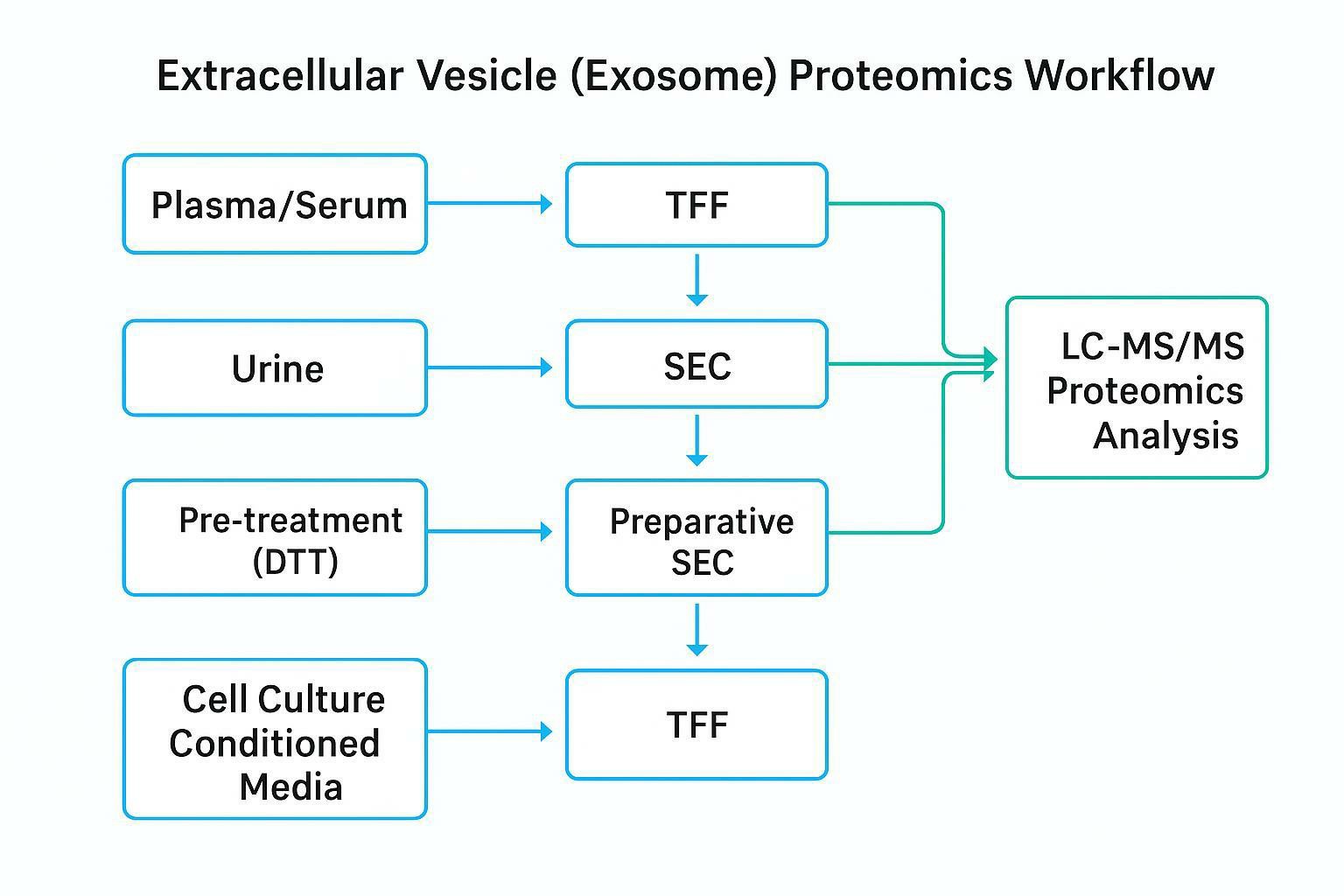
Preferred Workflows: TFF→SEC and gUC
When the goal is deep, interpretable LC–MS/MS, two workflow families are especially useful because they build in separation logic rather than relying on a single pelleting step.
Workflow A (common MS pipeline): TFF → SEC
Tangential Flow Filtration (TFF) is best thought of as a scalable, gentle concentrator: it reduces volume efficiently and can be tuned by membrane MWCO. But TFF is not a polishing step—you still need a way to remove soluble proteins and lipoproteins.
That's where SEC performs the critical role. SEC separates particles from a large fraction of soluble protein background by elution behavior.
Practical implications for proteomics:
- Treat SEC as the "purity gate" step.
- Pool fractions based on your own column + matrix mapping (more on fraction logic in the urine section).
- Expect that high-lipid or hyperlipidemic samples may need additional orthogonal strategies; plasma is not forgiving.
If you need support building a plasma/serum workflow that's explicitly designed to reduce lipoprotein-driven MS interference, Creative Proteomics' Exosome Isolation and Purification Service can be used as an outsourcing path for high-purity preparations.
Workflow B (orthogonal deep discovery): density gradient ultracentrifugation (gUC)
Density-gradient ultracentrifugation (gUC) is often chosen as an orthogonal approach when the project objective is deep discovery profiling and the team can tolerate lower throughput.
What gUC gives you is not magic yield—it gives you an extra physical separation axis (density) that can help disentangle EVs from co-isolating species that look similar by size alone. For discovery-stage proteomics, that additional orthogonality can be valuable when combined with stringent downstream QC.
A useful mental model:
- Use TFF to handle volume.
- Use SEC to remove soluble proteins.
- Use gUC when you need an additional "purity lever" and are willing to trade time/throughput.
Urine Protocols: Managing the Uromodulin Network
Urinary EV (uEV) workflows fail differently than plasma workflows. The dominant confounder is often uromodulin (also called Tamm–Horsfall protein, THP): a highly abundant urinary glycoprotein that can polymerize and form networks that trap vesicles and co-isolate protein complexes.
Chemical Intervention and SEC Fraction Mapping
Step 1: Disrupt THP trapping without turning your prep into a chemical soup
A common, mild intervention is DTT treatment, used to reduce disulfide-mediated polymer structures and help release vesicles from the THP network.
Two practical cautions for proteomics-focused labs:
- Standardize the intervention. Over- or under-treatment can change what co-isolates and create batch effects.
- Plan for downstream cleanup. Any chemical intervention increases the importance of buffer exchange and MS-compatible sample prep later.
Step 2: Map SEC elution windows for urine—and pool like you mean it
If you run SEC on urine without mapping fractions, you can easily "average" EVs together with THP-rich protein fractions.
A practical SEC pooling workflow for urine:
1. Run a representative urine sample through your SEC column.
2. Collect fractions at a consistent volume.
3. Evaluate fractions with a minimal panel:
- particle concentration (NTA or equivalent)
- EV marker presence (e.g., CD63/TSG101/ALIX depending on your target)
- uromodulin/abundant protein signal (e.g., uromodulin immunoblot, or protein assay trend)
4. Pool early EV-rich fractions.
5. Discard later THP-heavy fractions even if they contain protein that "looks like content." For LC–MS, that late protein is often exactly what hurts you.
Pro Tip: Write down your column + matrix fraction map as a lab-facing SOP artifact. For urine, fraction selection is often the difference between an EV proteome and a uromodulin proteome.
Cell Culture Media: Scalability and Background Depletion
Conditioned media (CCM) is the matrix where "classic benchtop ultracentrifugation" most visibly breaks—because the workflow has to process liters, not milliliters.
Two realities define CCM EV isolation for proteomics:
- You're usually dealing with large starting volumes.
- Your background is heavily shaped by culture inputs (especially serum supplementation).
Media Design and EV-Depleted Inputs
If your media contains serum, the experiment starts with a contamination problem before you ever harvest.
Two practical options are common:
- Transition to serum-free conditions when biologically feasible.
- Use rigorously validated EV-depleted FBS when serum is required.
The key is validation, not the label. EV-depleted inputs vary by preparation method, lot, and handling. If you do not verify the input background (particles and protein), you can end up measuring your supplement.
TFF→SEC for Large-Volume Processing
For CCM, the TFF→SEC logic becomes an engineering solution:
- Why batch ultracentrifugation fails at scale: It is time-intensive, capacity-limited, and often yields variable pellets across runs. For large volumes, you can also increase co-isolation of non-EV material simply by forcing massive quantities through a pelleting step.
- Why TFF works: It concentrates liters quickly with controllable parameters and is compatible with scale-up.
- Why SEC is still needed: Concentration alone doesn't remove soluble proteins. SEC is the polishing step that restores LC–MS friendliness.
A useful comparison table (high-level):
| Matrix | Dominant contaminant risk | Scalability bottleneck | Purity-forward backbone |
|---|---|---|---|
| Plasma/serum | Lipoproteins + abundant proteins | Contaminant overlap in size/density | TFF→SEC; add gUC for deep discovery |
| Urine | Uromodulin/THP network + protein complexes | Fraction selection and THP control | DTT (mild) → SEC with mapped pooling |
| Conditioned media | Serum-derived particles + supplement proteins | Processing liters reproducibly | TFF→preparative SEC |
Decision Framework: Aligning Isolation with Study Goals
EV proteomics projects usually fail when the isolation method is chosen for convenience rather than aligned with the study's translational stage.
A decision framework helps you avoid two common traps:
- Discovery trap: using a high-throughput but contaminated method and then concluding "there's no biomarker signal."
- Validation trap: using a deep, low-throughput method when you actually need consistent, scalable processing across many samples.
Discovery vs. Validation Priorities
Discovery Proteomics (maximum depth and interpretability)
- Goal: identify candidate biomarkers and pathways.
- Priority: purity and orthogonal separation.
- Typical backbone: SEC-based workflows; often TFF→SEC; add gUC when the matrix is exceptionally complex.
Targeted Validation (higher throughput, consistent selection)
- Goal: confirm a defined panel with targeted MS (PRM/MRM) or orthogonal assays.
- Priority: reproducibility and sample count.
- Typical backbone: higher-throughput enrichment (including capture approaches) paired with strong QC and acceptance thresholds.
LC-MS Compatibility and Pre-Digestion QC
Even strong isolation can be undone right before MS if buffers and QC gates aren't designed for proteomics.
Sample Prep and Cleanup
Buffer exchange is not optional when your upstream workflow introduces non-volatile salts, polymers, or reagents that suppress ionization or interfere with digestion. The safest way to avoid LC–MS surprises is to plan a consistent cleanup step before lysis and trypsin digestion.
For lysis, the general principle is MS-compatibility:
- Avoid harsh detergents like SDS unless you have a downstream cleanup workflow designed to remove them.
- Use denaturants and surfactants that are compatible with your digestion and cleanup strategy.
A deeper MS-centric overview of EV protein analysis is summarized in A guide to mass spectrometric analysis of extracellular vesicle proteins (2021).
Quality Metrics
A practical pre-MS QC gate typically combines particle measurement with protein burden.
Two quick checks used widely in EV workflows:
- Particle concentration and size distribution (e.g., NTA) to ensure the expected EV-enriched profile.
- A simple purity ratio concept: particles per microgram of protein.
The ratio is not a universal truth (different matrices behave differently), but it is a powerful trend metric: if particles stay flat while protein climbs, you're often concentrating soluble background rather than EVs.
Strategic Call-to-Action Integration
Scaling exosome isolation while maintaining MS-grade purity is a bioengineering problem: it requires matrix-aware preprocessing, separation that actually removes confounders (not just concentrates them), and QC gates that prevent low-quality inputs from reaching the instrument.
If you'd rather offload the workflow design and execution, Creative Proteomics' integrated subcellular platform can support custom TFF→SEC pipelines and end-to-end EV proteomics workflows—starting from isolation through LC–MS/MS readout via Extracellular Vesicles Proteomics Services and Mass Spectrometry Based Proteomics.
Frequently Asked Questions
Why is TFF followed by SEC the gold standard for large-volume EV proteomics?
TFF makes large-volume processing practical because it concentrates liters of conditioned media with controllable parameters and less batch-to-batch variability than repeated ultracentrifugation. SEC is then required to remove soluble protein background that otherwise suppresses LC–MS/MS signals, so the combined workflow improves both scalability and MS interpretability.
Which SEC fractions contain exosomes—and how do I choose the pooling window for proteomics?
EVs typically elute in early fractions (near the column void volume), while most soluble proteins elute later; the exact fraction numbers depend on the column and fraction volume. For proteomics, choose the pooling window by mapping a representative sample using particles (NTA), EV markers, and a readout for abundant contaminants (e.g., albumin in plasma or uromodulin in urine), then pool only the EV-rich early fractions and exclude protein-heavy late fractions.
How can I reduce lipoprotein contamination in plasma exosome isolation?
Start with strict pre-analytics (consistent pre-clear steps to reduce platelets and debris), then use a workflow with an explicit separation step rather than a single pellet. SEC-based purification is commonly used to reduce soluble protein background, and adding an orthogonal separation axis (e.g., density gradients or specialized chromatography) can further decouple EVs from lipoproteins that overlap in size and density.
What is uromodulin, and why does it cause problems for urinary EV proteomics?
Uromodulin (Tamm–Horsfall protein) is a highly abundant urinary protein that can polymerize into networks that trap vesicles and co-isolate protein complexes. In LC–MS/MS, this increases background protein burden and can mask low-abundance EV proteins—so THP control (pretreatment plus SEC fraction selection) is often central to successful urinary EV proteomics.
What lysis buffer is safe for isolated exosomes before LC-MS?
Use an MS-compatible lysis approach that solubilizes membrane proteins without leaving ion-suppressing reagents in the final digest. Avoid SDS unless your workflow includes a validated detergent-removal cleanup; many proteomics workflows use urea-based denaturation or MS-cleavable/compatible surfactants paired with a cleanup step and buffer exchange before LC–MS/MS.
Is ultracentrifugation still the best method for exosome isolation for proteomics?
It can be useful, but "best" depends on your matrix and goal. For discovery proteomics, adding SEC (and in some cases density gradients) is often favored because it reduces soluble background and improves interpretability; ultracentrifugation alone can co-isolate abundant non-EV material, especially from complex biofluids.
References
- A guide to mass spectrometric analysis of extracellular vesicle proteins
- A Review of Exosomal Isolation Methods: Is Size Exclusion Chromatography the Best Method for Exosome Isolation?
- Comparative analysis of tangential flow filtration and ultracentrifugation for the isolation of small extracellular vesicles from human plasma
- Protein Complexes in Urine Interfere with Extracellular Vesicle Biomarker Studies
- Urinary extracellular vesicles: a position paper by the Urine Task Force of the ISEV
- Uromodulin and the study of urinary extracellular vesicles
* For Research Use Only. Not for use in diagnostic procedures.
