
Proteomics aims to analyze the entire proteome encoded by the genome, reflecting cellular, organ, and organismal physiological changes through quantitative and qualitative changes in proteins. Post-transcriptional regulation and post-translational modifications result in a protein count far exceeding the number of genes.
With the rapid development of mass spectrometry techniques, mass spectrometry has gradually become the mainstream technology for proteomic analysis. Protein identification can be achieved by recognizing the unique peptide sequences, and mass spectrometry (MS) is a highly suitable technique for protein analysis due to its scanning speed, sensitivity, and ability to analyze complex mixtures.
The basic principle involves digesting proteins with enzymes like trypsin, separating the digested peptides using liquid chromatography (LC), and detecting the separated peptide ions using matrix-assisted laser desorption/ionization (MALDI) or electrospray ionization (ESI). The peptide ions are then ionized and eluted in liquid chromatography, and their molecular weights are determined in the mass spectrometer. The mass spectrometer further analyzes the composition of the fragments.
The entire workflow of liquid chromatography and mass spectrometry is commonly referred to as tandem mass spectrometry (MS/MS). Tandem mass spectrometry identifies specific amino acid sequences, and with the analysis using protein databases and software, qualitative and quantitative results of proteins can be obtained.
Proteomic research depends on experimental techniques, high-resolution and high-sensitivity mass spectrometry, as well as quantitative algorithms and software updates, providing the means and technical assurance for quantitative proteomics.
Traditional protein identification methods, such as immunoblotting, chemical sequencing, and gel electrophoresis, are generally time-consuming, labor-intensive, and low-throughput, making them unsuitable for high-throughput proteomics studies. The advent of mass spectrometry has expanded the application of traditional techniques, such as the combination of two-dimensional gel electrophoresis and mass spectrometry (2DE with MALDI-TOF/MS).
As mass spectrometry technology advances, various non-two-dimensional gel electrophoresis separation techniques have emerged, including multidimensional liquid chromatography-based protein identification technology (MudPIT), top-down mass spectrometry, and sub-proteomics.
Stable isotope labeling methods for samples are typically categorized as labeled quantitative and label-free methods.
Select Service
Labeled Quantitative Proteomics
Isotope-Coded Affinity Tag (ICATs)
Isotope-Coded Affinity Tag (ICAT) is a technique that utilizes ICAT reagents for protein separation. It involves labeling cysteine residues in proteins from different cells with isotopically distinct affinity tags (ICATs) and using tandem mass spectrometry for mass spectrometric analysis of complex samples.
The key to the ICAT method lies in the practical application of the ICAT reagent, which consists of three parts. The middle part, known as the linker, contains either 8 hydrogen atoms (light chain reagent) or deuterium atoms (heavy chain reagent). One end of the linker is attached to a thiol-reactive group that can react with the thiol group of cysteine residues in proteins, facilitating protein labeling. The other end is linked to biotin, used for affinity purification of labeled proteins or peptide chains.
When using ICAT technology for protein separation and quantification, proteins are first labeled, followed by enzymatic digestion and affinity chromatography separation of the digested peptide complex. Only the peptides labeled with isotopes can enter and be retained by the chromatographic column for mass spectrometric identification. By comparing the signal intensities of heavy and light chain reagent peptides in mass spectrometry, quantitative analysis of differentially expressed proteins can be achieved.
Advantages of ICAT include its ability to directly analyze mixed samples from normal and diseased cells or tissues, enabling rapid qualitative and quantitative identification of low-abundance proteins, especially hydrophobic proteins such as membrane proteins. It can quickly identify important biomarkers involved in diseases. ICAT can be used not only for analyzing the entire cellular proteome but also for quantifying and detecting specific subcellular components, including those in mitochondria.
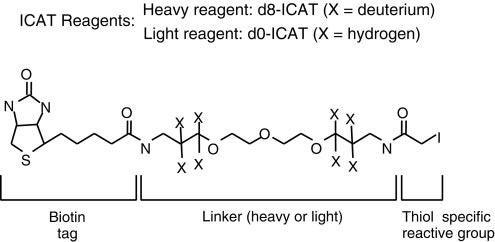 Isotope-coded affinity tags (ICAT) (Kenyon et al., 2020).
Isotope-coded affinity tags (ICAT) (Kenyon et al., 2020).
Enzyme-Catalyzed 18O Isotope Labeling
Under typical conditions in the presence of 18O, enzymatic digestion of proteins can generate peptides labeled with isotopes. The most commonly used approach is to introduce 18O during the process of trypsin digestion. Alternatively, intermediate endopeptidases such as Glu-C and Lys-C can be employed to introduce an oxygen atom during peptide cleavage reactions, achieving isotope labeling.
Isobaric Tags for Relative and Absolute Quantification (iTRAQ)
The principle of Isobaric tags for relative and absolute quantification (iTRAQ) involves enzymatic digestion followed by isotope labeling of amino-terminal and lysine residues of peptides.
iTRAQ reagents consist of three chemical groups: a reactive group, a reporter group, and a balance group. The reactive group specifically reacts with the amino groups of peptides (at the N-terminus and lysine residues). The reporter group has different mass numbers, and with the addition of the corresponding balance group, the total mass of the iTRAQ reagent remains constant. This ensures that peptides labeled with different tags have the same retention time in chromatography.
Due to their equal mass, peptides labeled with different tags appear as a single precursor ion peak in the first-level spectrum. Upon further fragmentation, the balance group undergoes neutral loss, while the reporter group generates corresponding reporter ions with different mass numbers. By comparing the abundance of reporter ions, relative abundance ratios of samples can be obtained. Currently, iTRAQ allows simultaneous labeling and quantification of 4 or 8 samples.
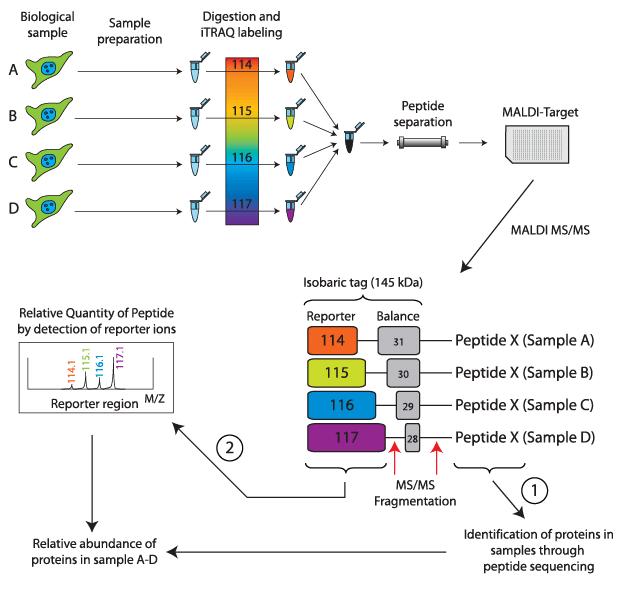 Basic principle of the iTRAQ labeling technology (Pernemalm et al., 2009).
Basic principle of the iTRAQ labeling technology (Pernemalm et al., 2009).
Tandem Mass Tag (TMT)
TMT (Tandem Mass Tag) technology is a peptide in vitro labeling technique developed by Thermo Fisher Scientific in the United States.
The technology utilizes isotopic labels such as 6-plex (TMTsixplex™ Isobaric Label Reagent Set), 10-plex (TMT10plex™ Isobaric Label Reagent Set), and 16-plex (TMTpro 16plex) to selectively label peptides from different sources by connecting to specific amino acid sites. Subsequently, tandem mass spectrometry analysis is conducted to monitor the labeled fragments, enabling quantitative analysis of peptides.
In a single experiment, TMT technology allows flexible comparison of the relative protein content in up to 16 different samples. In TMT experiments, total proteins are extracted from tissue or cellular samples, enzymatically digested into peptides, and then labeled with TMT reagents.
TMT reagents consist of three main parts: a reporter group, a balance group, and a peptide reactive group. Taking the 6-plex reagent as an example, the reporter groups have relative molecular masses of 126, 127, 128, 129, 130, and 131, while the balance groups have relative molecular masses of 103, 102, 101, 100, 99, and 98. This results in six isobaric tags with an equal molecular mass of 229.
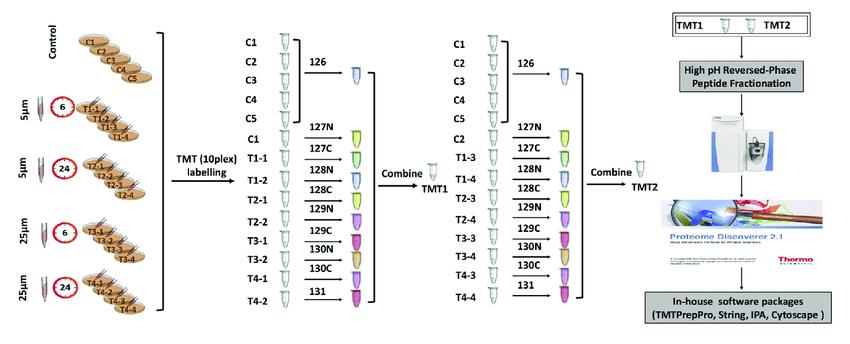 Tandem mass tag (TMT) labeling strategies and experimental workflow (Deng et al., 2019).
Tandem mass tag (TMT) labeling strategies and experimental workflow (Deng et al., 2019).
Label-Free Quantitative Proteomics
Mass spectrometry-based labeling for quantitative proteomics applications is widely used. However, the drawbacks include the high cost of labeling reagents and the complexity of sample preparation, limiting the repeatability in handling complex samples.
The solution to this problem comes in the form of label-free quantification technology, which directly utilizes MS signals for quantification. Label-free techniques do not require expensive labeling reagents, making them suitable for quantitative analysis of various sample types. They are extensively applied in quantitative proteomics research.
Label-free quantitative proteomics based on mass spectrometry can be broadly categorized into two principles: one is based on the quantification algorithm of Extracted Ion Current (XIC), and the other is based on Spectral Counting (SC) for relative quantification of protein peaks.
Extracted Ion Current (XIC): This method primarily involves extracting the signal intensity corresponding to different retention times on the retention time (RT) axis based on the mass-to-charge ratio of the peptide precursor ion. The XIC area or the sum of signal intensities is used as the quantitative result for the peptide.
To accurately calculate the corresponding XIC peptide sequence, two approaches are commonly employed: 1) matching with an Accurate Mass and Time (AMT) tag database model, and 2) using the results of a protein sequence database search to reverse-determine the corresponding peptide sequence.
Spectral Counting (SC): According to mass spectrometry principles, the higher the abundance of a protein, the higher the abundance of enzymatically cleaved peptide segments, resulting in a higher probability of detection by mass spectrometry.
The number of spectra counting the peptides identified for a specific protein is used as a measure of protein abundance. The spectral counting method is simple, intuitive, and convenient for calculation, making it widely used in label-free quantitative methods.
For label-free quantitative proteomics, different mass spectrometry data acquisition modes can be chosen based on experimental needs, such as Data-Dependent Analysis (DDA) and Data-Independent Analysis (DIA).
DDA is based on the shotgun proteomics principle, collecting precursor ions for fragmentation in the order of decreasing ion intensity. This approach has some randomness and intensity dependence in its results.
DIA was first proposed in 2000 and divides the entire scan range into multiple windows, continuously and unbiasedly collecting information for all MS2, maximizing the acquisition of fragment ion information to improve quantitative accuracy.
In 2012, a method combining SWATH and DIA was introduced, achieving rapid high-throughput analysis of large sample queues through the use of high-resolution mass spectrometry and optimized acquisition schemes. For the latest developments in label-free quantitative proteomics, Direct DIA (Direct data independent analysis) and 4D-DIA (adding ion mobility dimension information) methods have been proposed, aiding in the quantitative analysis of proteomics and post-translational modifications.
References
- Kenyon, George L., et al. "Defining the mandate of proteomics in the post-genomics era: workshop report." Molecular & Cellular Proteomics 1.10 (2002): 763-780.
- Pernemalm, Maria. Cancer proteomics: Method development for mass spectrometry based analysis of clinical materials. Karolinska Institutet (Sweden), 2009.
- Deng, Liting, et al. "Amyloid β induces early changes in the ribosomal machinery, cytoskeletal organization and oxidative phosphorylation in retinal photoreceptor cells." Frontiers in Molecular Neuroscience 12 (2019): 24.
Related Services

* For Research Use Only. Not for use in diagnostic procedures.
