
Immunoaffinity capture sits at an unusual intersection in EV workflows. It can deliver the specificity needed to answer cell- or disease-source questions, but it can also lock selection bias into your dataset if you treat a marker as a universal handle.
This resource is for decision-stage teams that need an enrichment strategy they can defend: what to capture, why those markers, what you might miss, and which controls make the conclusions credible.
When to use immunoaffinity capture for EV subpopulation enrichment
Immunoaffinity capture (often implemented as antibody-coated magnetic beads) enriches EVs based on surface epitopes rather than size or density. The advantage is simple: you can pull a defined subpopulation out of a complex background, which is hard to do with bulk methods alone.
It's most useful when your biological question is explicitly subpopulation-based, for example:
- You need a marker-defined EV subset for mechanistic work (receptor–ligand interactions, uptake assays, functional readouts).
- You are profiling EV cargo where contaminants are a major confounder (certain proteomics and targeted assays).
- You need to compare marker-positive fractions across conditions while keeping the rest of the matrix constant.
Common sample types where immunocapture can be effective include plasma and serum (high protein background), cerebrospinal fluid (low particle abundance, high sensitivity requirements), urine, and conditioned media. In these matrices, a practical decision is often to do a bulk clean-up first and then use immunocapture for subpopulation definition, rather than trying to capture directly from raw input.
If your starting point is a complex clinical sample and your priority is to reduce matrix-derived interference before selecting a subpopulation, a workflow that begins with exosome isolation and purification (for example, by SEC or density-based steps) can make the affinity step more interpretable.
Key Takeaway: Immunoaffinity capture is a subpopulation definition tool. Use it when "which EVs?" matters more than "how many EVs?"
How to choose the right markers for EV immunocapture
Marker selection is where antibody-based exosome isolation succeeds or quietly fails. A good capture marker is (1) exposed on the vesicle surface, (2) present on the target subpopulation at usable density, and (3) informative for the biology you want to measure.
Key surface markers: CD63, CD81, CD9, and their applications in EV subpopulation enrichment
CD9, CD63, and CD81 are widely used tetraspanins associated with small EVs. In practice, they are often treated as "general" EV handles, but they are better understood as common markers with heterogeneous expression.
Use cases where each is often chosen:
- CD63: frequently used when the goal is to enrich for vesicles that fit canonical exosome marker expectations in downstream marker validation (for example, Western blot panels).
- CD81: often selected when single-particle profiling or co-expression analysis suggests CD81-high subfractions in a given biofluid or cell source.
- CD9: commonly used in many biofluids and cell culture contexts, but its utility depends on how enriched CD9 is in your source and whether your target subpopulation is CD9-positive.
If you are making CD63/CD81 EV marker selection decisions for a cohort, start with a baseline check: what fraction of particles in your input appear CD9/CD63/CD81-positive, and are those markers co-expressed or mutually exclusive in your system?
Multiplex capture strategies and selecting the best marker panel
If your goal is EV subpopulation isolation techniques that remain stable across cohorts, multiplex capture is often safer than betting the study on a single tetraspanin.
Two common multiplex patterns:
- "Coverage" panels: combine CD9/CD63/CD81 to reduce the chance that a target-negative fraction dominates the biological signal.
- "Specificity" panels: combine a tetraspanin with a disease- or tissue-associated marker to enrich a more interpretable subset.
A practical way to decide is to formalize the trade-offs in a table before you buy antibodies, beads, and sample volume.
| Marker strategy | What it captures well | Main bias risk | When it's a strong choice |
|---|---|---|---|
| Single tetraspanin (CD63 or CD81 or CD9) | Clean, marker-positive fraction | Underrepresents marker-low/negative EVs; fragile cross-sample comparability | Small pilot studies, method development, single-source samples |
| Multi-tetraspanin (CD9+CD63+CD81) | Broader sEV coverage | Still biases away from non-tetraspanin subtypes; antibody competition possible | Cohorts, mixed sample types, discovery proteomics |
| Tetraspanin + tissue/disease marker | Biologically targeted subset | Strong selection bias by design; depends on marker validity | Targeted biomarker programs, mechanistic source attribution |
Tissue and disease-specific markers for targeted EV isolation
If you need targeted EV isolation, tissue and disease-specific markers can be powerful. They also raise the burden of proof.
Before you capture, define what would convince a skeptical reviewer that the marker tracks the source you claim:
- Evidence that the marker is surface-accessible on EVs in your matrix.
- Evidence that the marker is not dominated by soluble forms or non-vesicular complexes in that matrix.
- An orthogonal readout showing enrichment of expected cargo or functional behavior.
For downstream EV immunocapture methods for proteomics, be careful with markers that co-purify abundant plasma proteins or immunoglobulins. Those can inflate identification counts while reducing interpretability.
How marker heterogeneity affects EV enrichment
Marker heterogeneity is not a small nuisance; it is a defining property of EV populations. A key implication is that "CD63-captured EVs" are not interchangeable with "CD81-captured EVs," and neither is a neutral proxy for "all EVs."
In human biofluids, CD9/CD63/CD81 profiles can vary by sample type, with distinct patterns across serum/plasma compared with CSF and brain-adjacent material. When you move between matrices, disease settings, or tissue-derived preparations, a single-marker capture can shift from "reasonable proxy" to "systematic blind spot" without warning.
Variability in marker expression across different sample types
In practical terms, variability shows up as:
- Different dominant tetraspanin signals depending on matrix and tissue proximity.
- Shifts in co-expression patterns that change how well a multiplex panel behaves.
- Increased epitope masking in high-protein backgrounds that reduces capture efficiency without changing true EV abundance.
Why selecting the right combination of markers is crucial for accurate subpopulation capture
If you are comparing cases vs controls, or responders vs non-responders, the central question is whether your capture step is stable across groups.
A good marker combination does two things:
- It captures the subpopulation you claim.
- It fails in a way you can detect (via controls and post-capture profiling), rather than failing silently.
This is where "best markers for EV enrichment" becomes a system-specific question, not a universal answer.
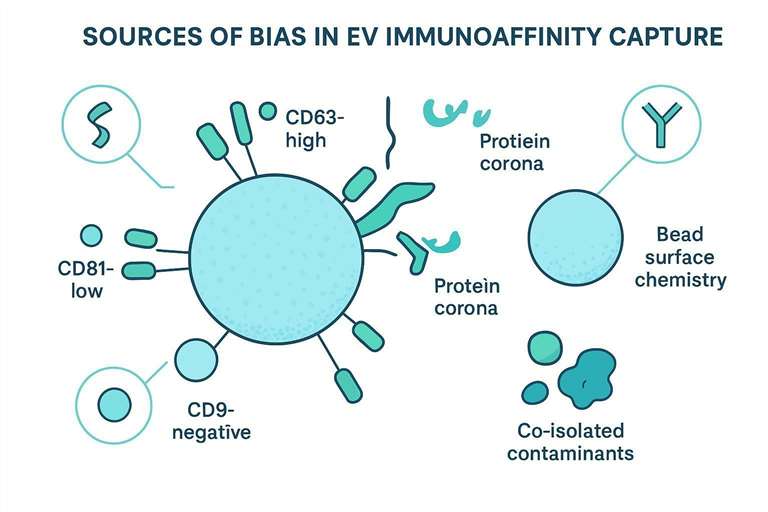
Bias in EV immunocapture: sources and implications
Bias in EV immunoaffinity capture can enter at multiple layers. Some are biological (what the cells release). Some are technical (what your antibodies and surfaces prefer). Some are pre-analytical (what your sample handling changed).
Biological heterogeneity and underrepresentation of certain EV subpopulations
Immunocapture enriches what it can bind. EVs lacking the target epitope are absent by design. That is fine if your claims are explicitly limited to the captured population, but it becomes a problem when readers infer you measured "the EVome" of the sample.
Two common failure modes:
- Your disease group shifts marker expression, so capture yield changes for biological reasons unrelated to total EV abundance.
- Your target subpopulation is rare and variable, so stochastic sampling dominates the measured signal.
Antibody-specific and surface chemistry biases in immunoaffinity capture
Antibody clones differ. So do immobilization strategies, bead materials, and antibody density. These differences can bias toward:
- EVs with higher epitope density.
- EVs where the epitope is more exposed (orientation effects).
- EVs that nonspecifically adsorb to the surface chemistry you chose.
If you can't elute intact EVs cleanly (because the linkage chemistry is effectively irreversible in practice), you may also bias which downstream assays are feasible.
Pre-analytical biases and co-isolated contaminants
Pre-analytical variability often determines whether "how to avoid bias in EV immunocapture" is achievable in your setting.
Examples that commonly matter:
- Serum vs plasma choice and anticoagulant effects.
- Freeze–thaw cycles and storage duration.
- Clotting-derived particles and protein aggregates.
- Lipoproteins and abundant proteins that either co-isolate or mask epitopes.
Even marker readouts can mislead if you use poor proxies. Acetylcholinesterase activity has been used as a generic EV marker in some contexts, but evidence indicates it can reflect serum-derived non-vesicular components rather than EV abundance. Treat it cautiously in purity assessment.
⚠️ Warning: If your purity metric is correlated with serum exposure or matrix carryover, it can "validate" a contaminated preparation.
Designing effective controls for EV immunoaffinity capture
Controls should tell you three things: what fraction is nonspecific, what fraction is missed, and whether the captured material behaves like vesicles.
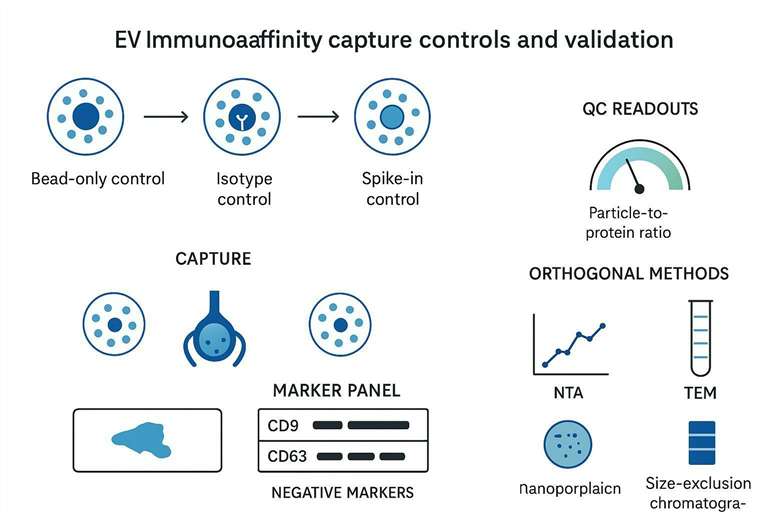
Negative and mock controls: bead-only and isotype controls
For immunoaffinity EV isolation, two negatives are basic but non-negotiable:
- Bead-only control: incubate the same sample with beads lacking capture antibody to estimate nonspecific adsorption.
- Isotype control: use a matched isotype antibody to quantify antibody-driven nonspecific binding.
Report these controls as quantitative readouts (particles, protein, or signal intensity), not as "performed" checkboxes.
Orthogonal validation with baseline samples and spike-ins
If your goal is subpopulation enrichment, you need a baseline profile of the input.
Practical options:
- Measure input EV marker distribution (single-particle phenotyping where possible).
- Use spike-ins that behave like EVs for process control, focusing on recovery consistency across batches.
- Compare the captured fraction and the flow-through fraction to demonstrate selective enrichment rather than generalized depletion.
Post-capture controls: purity and functional verification
Post-capture verification is where many studies stop too early.
A strong post-capture package includes:
- Evidence of expected EV markers in the captured fraction.
- Evidence of depletion of common contaminants (negative markers).
- A functional sanity check when the downstream assay depends on intact vesicles (uptake, signaling, enzyme activity), using conditions that do not introduce artifacts.
Purity and specificity validation for immunocaptured EVs
Validation needs to match what you plan to claim. If you claim a marker-defined EV fraction is enriched, show enrichment. If you claim you have "pure EVs," show purity with more than one metric.
Evaluating purity: particle-to-protein ratio and marker validation
Particle-to-protein ratio can be a useful screening metric, but it is not definitive on its own. Combine it with marker validation.
A practical validation panel often includes:
- Canonical positive EV-associated markers (commonly tetraspanins in many settings).
- Negative markers for cellular compartments or non-vesicular contaminants.
- A check for matrix-specific confounders (for example, abundant lipoprotein-associated proteins in plasma-derived preparations).
If you need a standardized characterization package with NTA, TEM, and marker panels, exosome analysis services can serve as a reference for what a complete post-capture validation bundle typically includes.
Ensuring functional integrity for downstream assays
Immunocapture can preserve vesicle integrity, but harsh elution and aggressive washing can damage surface proteins or strip loosely associated cargo.
If your downstream assay is functional (uptake, receptor activation), design a release strategy and controls that test whether bead-bound EVs behave differently from released EVs.
Orthogonal validation methods: NTA, TEM, and Western blot
Orthogonal methods answer different questions:
| Method | What it tells you | What it does not guarantee |
|---|---|---|
| NTA | Particle concentration and size distribution trends | That particles are EVs (protein aggregates can still appear) |
| TEM | Vesicle-like morphology and qualitative purity | Quantitative yield; marker identity |
| Western blot | Presence/absence of selected markers | Single-vesicle heterogeneity; non-vesicular complexes sharing epitopes |
Use at least two orthogonal readouts when the data will be used for decision-making in a biomarker or mechanistic program.
Combining immunoaffinity capture with other EV isolation methods
Combining methods is often how teams reconcile purity and yield.
Integrating SEC, ultracentrifugation, and immunoaffinity for comprehensive EV profiling
A common pattern for complex samples is:
- Bulk enrichment and cleanup (SEC and/or differential ultracentrifugation, depending on the matrix and throughput needs).
- Immunoaffinity capture to define the subpopulation.
- Post-capture validation and downstream omics or functional assays.
This sequence reduces epitope masking and nonspecific adsorption before you ask antibodies to do fine selection.
For teams that plan downstream protein identification and quantification after capture, exosome proteomics services can be a practical next step, since proteomics workflows are particularly sensitive to co-isolated contaminants and batch effects.
When to combine methods for enhanced purity and yield
Combine methods when:
- The matrix is highly complex (plasma/serum) and direct capture yields inconsistent results.
- You need both a "total EV" baseline and a marker-defined subset.
- You plan discovery proteomics where contaminant carryover can dominate identifications.
If the project spans EV types beyond a single small-EV subset, extracellular vesicles proteomics services may be more aligned with broader profiling needs.
Practical considerations for immunoaffinity capture workflows
A reliable protocol for immunoaffinity EV isolation is rarely about one magic incubation step. It's about controlling contact time, minimizing nonspecific adsorption, and choosing elution conditions that preserve what you care about.
Sample preparation, antibody incubation, and washing steps
Key implementation details worth standardizing:
- Pre-clear strategy (low-speed spins, filtration, or SEC) to reduce debris and high-abundance background.
- Incubation conditions (temperature, mixing, time) that are consistent across batches.
- Bead-to-sample ratio and antibody density; document lot numbers when possible.
- Wash stringency that removes background without stripping weakly bound vesicles.
Pro Tip: If yields vary batch-to-batch, check whether the input marker profile changed (sample handling) before blaming capture chemistry.
Optimizing elution conditions and minimizing contaminant co-precipitation
Elution is a decision: do you need intact vesicles off the beads, or is bead-bound analysis acceptable?
- If you need intact EVs, prioritize release conditions that preserve surface proteins and avoid conditions that promote aggregation.
- If you don't need intact EVs, you can focus on assay compatibility (for example, direct lysis for proteomics) while still controlling nonspecific binding.
Regardless, keep contaminants in mind. Many matrices contain components that bind both antibodies and bead surfaces. The bead-only and isotype controls are your early warning.
Reporting standards for immunoaffinity EV capture
Decision-stage readers need reporting that makes results reproducible and comparable.
Essential reporting for reproducibility and transparency
At minimum, report:
- Sample type and handling: anticoagulant (if applicable), storage, freeze–thaw history.
- Pre-processing steps (spins, filtration, SEC) with key parameters.
- Capture details: antibody clone, bead type, antibody density/amount, incubation conditions.
- Wash and elution conditions.
- Controls: bead-only, isotype, and any spike-in or baseline profiling.
- Characterization methods and markers (positive and negative panels).
MISEV and EV-TRACK guidelines for standardized reporting
MISEV guidelines define minimal expectations for EV studies, including characterization and transparent reporting. EV-TRACK complements this by emphasizing method transparency and standardized reporting fields that make cross-study comparisons less fragile.
Key parameters to report for cross-study comparability
A compact checklist for publications and internal method transfer:
| Category | Parameters that most often break comparability |
|---|---|
| Sample | serum vs plasma, anticoagulant, storage, freeze–thaw |
| Pre-clear | filtration pore size, SEC fraction choice, spin times |
| Capture | clone, bead chemistry, incubation time/temperature, mixing |
| Wash/elute | buffer composition, stringency, release approach |
| Readouts | marker panel choice, thresholds, instrument settings |
Best practices for EV immunoaffinity subpopulation studies
A subpopulation paper that survives scrutiny usually does the same few things well.
Summary of key points: marker choice, bias mitigation, control design
- Choose markers based on baseline profiles in your matrix, not on generic assumptions.
- Prefer multiplex approaches when comparing across heterogeneous sample types.
- Treat bias as a measurable variable: quantify nonspecific capture and report it.
- Validate with orthogonal readouts, and be explicit about what the captured fraction represents.
Practical steps for improving reproducibility and minimizing experimental bias
If you want one operating model:
- Pre-clear the matrix enough to reduce epitope masking.
- Profile the input marker distribution to select a marker panel.
- Run bead-only and isotype controls every batch.
- Compare captured vs flow-through fractions.
- Validate with at least two orthogonal methods.
- Report parameters with enough detail that another lab could reproduce the capture.
FAQs
How does immunoaffinity capture differ from ultracentrifugation or SEC for EV isolation?
Immunoaffinity capture enriches EVs based on specific surface epitopes, while ultracentrifugation and SEC primarily separate by physical properties (size/density). Immunocapture is the better fit when you need a marker-defined subpopulation; bulk methods are better when you need a broad EV fraction for baseline profiling.
What are the best markers for EV enrichment: CD63, CD81, or CD9?
There isn't one best marker across all sample types. CD9/CD63/CD81 expression can vary by biofluid and tissue source, so a marker that captures well in plasma may miss a major fraction in CSF or tissue-adjacent samples. For cross-sample studies, multiplex panels (e.g., CD9+CD63+CD81) often reduce the risk of single-marker bias.
How can I avoid bias in EV immunocapture when comparing disease vs control cohorts?
Start by profiling marker distributions in a subset of input samples from both groups, then choose a capture panel that remains stable across conditions. Run bead-only and isotype controls per batch, and report captured and flow-through fractions so readers can see what was excluded.
Why can immunoaffinity capture yield look low even when EV abundance is high?
Yield reflects the fraction of EVs that present an accessible copy of the target epitope under your incubation conditions. Protein corona effects, soluble competitors, and epitope orientation can all reduce apparent capture without changing the true particle count.
What quality control in immunoaffinity EV isolation is most convincing for reviewers?
Show orthogonal evidence: particle trends (NTA), vesicle morphology (TEM), and marker enrichment/depletion (Western blot or immunoassays with positive and negative markers). Report nonspecific binding from bead-only and isotype controls so purity claims are grounded.
Can I do EV immunocapture and still run proteomics on the captured fraction?
Yes, but plan for it early. Proteomics is sensitive to co-isolated proteins (especially from plasma/serum), so pre-clear steps and stringent controls matter. Also decide whether you will elute intact EVs or lyse on-bead, and keep that decision consistent across samples.
References
- Tetraspanin heterogeneity of small extracellular vesicles in human biofluids
- Acetylcholinesterase is not a generic marker of extracellular vesicles
- Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines
- Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches
* For Research Use Only. Not for use in diagnostic procedures.
