
Therapeutic extracellular vesicles (EVs), including exosomes, are increasingly being evaluated as delivery vectors for nucleic acids, proteins, and gene editors. Not because they're trendy, but because they solve problems that synthetic carriers still struggle with: native biocompatibility, lower innate immunogenicity in many contexts, and the ability to display biologically meaningful surface proteins that influence circulation time and tissue interactions.
This therapeutic potential comes with a tradeoff: EVs are biological products with inherent heterogeneity. For EV-enabled therapies, CD63 detection and a particle-size profile alone are insufficient for therapeutic EV characterization. IND-enabling development requires a Chemistry, Manufacturing, and Controls (CMC) package that treats EVs as complex, multi-component drug products whose quality must be controlled through a defensible Critical Quality Attribute (CQA) matrix.
The CMC Paradigm Shift: Establishing Critical Quality Attributes (CQAs) for Exosome Drug Delivery
Synthetic lipid nanoparticles (LNPs) changed what was possible for nucleic acid delivery, and they did it with a manufacturing mindset that most regulators already know: defined inputs, tightly controlled formulation, and relatively compact composition. Therapeutic EVs flip that model. They're secreted by living cells and can carry a dense and variable surfaceome, lipidome, and proteome that all influence performance.
For developers, the shift isn't philosophical. It's practical. The characterization plan that's "good enough" for academic claims won't survive regulatory scrutiny when you're trying to prove identity, purity, and potency under CMC.
Why Conventional EV Characterization Is Insufficient
Nanoparticle Tracking Analysis (NTA) provides particle size distribution and particle concentration. Western blotting for CD63/CD81 can support the presence of EV-associated proteins. But neither approach can:
- distinguish EVs from similarly sized contaminants (protein aggregates, lipoproteins, cell debris)
- confirm cargo is truly intravesicular vs surface-adhered
- quantify key safety risks (e.g., procoagulant proteins)
- demonstrate batch-to-batch comparability at scale
Guidance documents such as the International Society for Extracellular Vesicles' updated recommendations in Minimal information for studies of extracellular vesicles (MISEV2023): updated guidelines of the International Society for Extracellular Vesicles (PubMed) are not regulatory filings, but they're a useful baseline for what "rigorous EV characterization" looks like when you need orthogonal evidence.
A practical CQA matrix for therapeutic EV development
A workable CQA framework ties each attribute to a patient risk and an analytical control strategy. In practice, that means using orthogonal assays for the same claim, and selecting assays that remain valid across scale-up.
| CQA domain | Attribute (what you must control) | Why regulators care (risk) | Examples of orthogonal evidence (how to test it) |
|---|---|---|---|
| Identity | EV markers and EV-associated protein profile | Mis-identified product; changing "product" over time | Immunoassay/Western (markers) + LC-MS/MS proteome fingerprint |
| Purity | Host cell proteins (HCP), non-EV proteins, debris | Immunogenicity, toxicity, procoagulant risk | Deep LC-MS/MS impurity map + targeted assays for high-risk proteins |
| Safety | Procoagulant/adventitious proteins | Thrombosis risk; unexpected inflammatory signals | Proteomics screening for candidates + functional coagulation readouts as needed |
| Cargo | Encapsulated drug amount per vesicle | Dose control; potency and PK variability | Protease protection + targeted MS quantification (PRM) |
| Heterogeneity | EV subpopulations and size distribution | Variable biodistribution and efficacy | Single-particle phenotyping + size distribution + proteome clustering |
| Potency | MoA-linked activity | Release decisions must link to function | Cell-based potency assay aligned to MoA + surface display / cargo QC |
For teams that need a single integrated characterization workflow (from isolation/phenotyping to multi-omics), Comprehensive Exosome Isolation, Characterization, Phenotyping, and Multi-Omics Services can be used to build an assay package that stays consistent across early discovery, process development, and comparability studies.
Purity and Safety: Profiling Host Cell Protein (HCP) Impurities
One of the central challenges in therapeutic EV CMC is not only demonstrating EV identity, but also defining and controlling co-isolated impurities. EVs are secreted by producer cells (MSC, HEK293T, or engineered lines), and the harvest contains a crowded background of proteins, lipids, nucleic acids, and membrane fragments. Even after downstream purification, co-isolated impurities can persist, and some are not just inconvenient; they're potentially hazardous.
The most defensible purity strategy is to build an impurity map early, then prove that downstream steps consistently remove those impurities without shifting the EV identity profile.
Deep LC-MS/MS for HCP and Procoagulant Clearance
Untargeted discovery proteomics (deep LC-MS/MS) is the most direct way to establish what your EV preparation contains beyond a short marker panel. In a CMC context, the output is not a long protein list. The output is a repeatable "fingerprint" that supports three claims:
- Identity stability: a consistent core EV-associated proteome across lots
- Impurity knowledge: a defined set of HCPs and other adventitious proteins that must be controlled
- Safety screening: the ability to proactively look for risk-associated proteins (for example, procoagulant factors)
In practice, developers often combine:
- a discovery layer (deep LC-MS/MS) to build the impurity map
- a targeted layer (selected PRM/MRM panels) for "must-control" proteins once risks are identified
For teams building MS-driven impurity and identity control strategies, extracellular vesicle proteomics services can support both discovery profiling and targeted validation (including PRM) as methods mature.
Demonstrating Batch-to-Batch Comparability
Batch-to-batch comparability is where EV heterogeneity becomes a regulatory risk. As you scale from flasks to bioreactors, or change filtration and chromatography conditions, you can unintentionally:
- shift EV subpopulation composition
- enrich different surface proteins
- co-isolate different impurity profiles
A strong comparability argument uses proteomic fingerprinting to show that:
- the core EV proteome is stable
- the impurity signature shrinks (or remains controlled) across downstream steps
- the process changes do not introduce new, high-risk contaminants
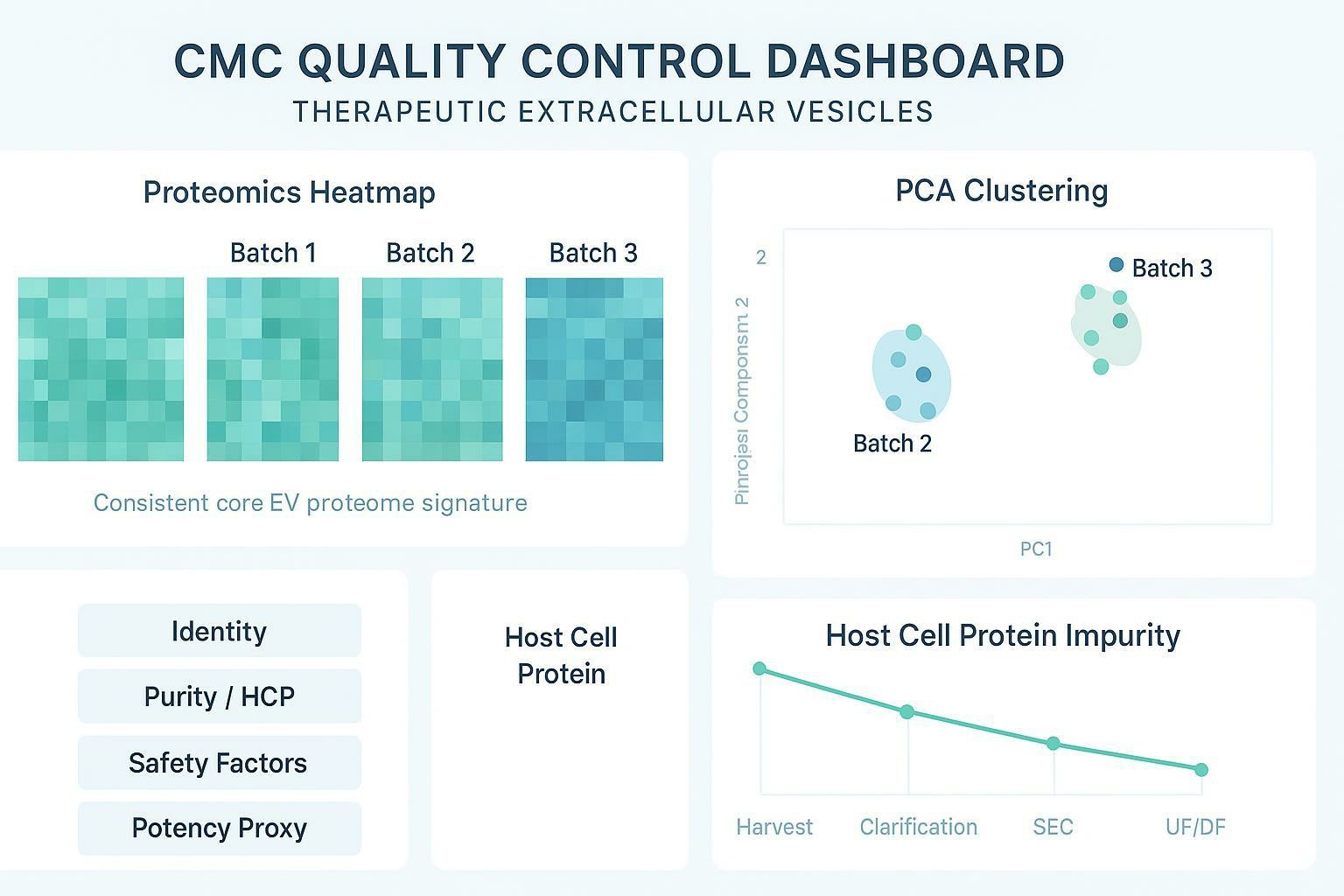
A useful way to communicate comparability internally and to regulators is a dashboard-style summary with orthogonal metrics. For example:
| Comparability question | Evidence that holds up under scale-up | What failure looks like |
|---|---|---|
| Did EV identity drift? | LC-MS/MS fingerprint + marker panel stability | PCA/cluster shift; new dominant proteins |
| Did purity improve or worsen? | Impurity map trend + targeted HCP panel | New impurity classes; rising HCP signatures |
| Did safety risk increase? | Screening for procoagulant/immune-active candidates + functional readouts when needed | Emergence of risk proteins or aggregates |
Cargo Quantification: Proving True Encapsulation
Cargo loading is a common source of overinterpretation in therapeutic EV development.
If your therapeutic is a protein, enzyme, or gene editor, the question isn't "did we detect it in the prep?" The question is "how much of it is actually protected inside intact vesicles?" Without that distinction, you can overestimate loading, mis-predict potency, and build PK expectations on artifacts.
Surface adsorption is common. Many proteins stick to membranes or co-aggregate with EVs during loading. If you don't control for that, an immunoblot or bulk MS signal can look like successful encapsulation.
The Protease Protection Assay and PRM Validation
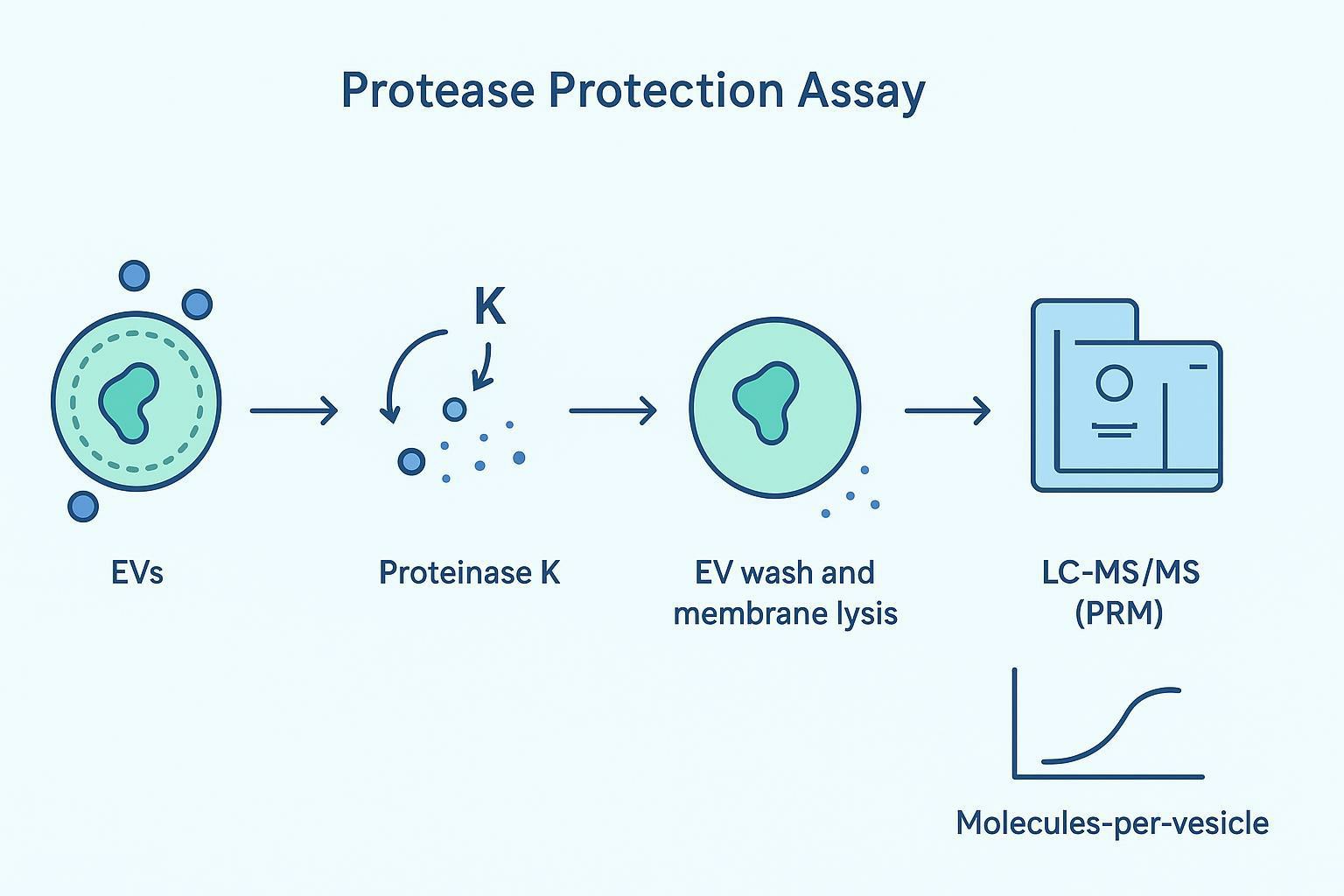
A protease protection assay is one of the clearest ways to separate intraluminal cargo from surface-adhered cargo.
Core logic: an intact EV lipid bilayer protects internal cargo from proteolysis. Anything exposed on the outside gets digested.
A regulator-facing workflow looks like this:
- Treat the loaded EV prep with an external protease (commonly Proteinase K) under conditions that digest exposed proteins.
- Remove the protease and digested fragments (wash/cleanup).
- Lyse the washed EVs.
- Quantify the protected therapeutic cargo using targeted mass spectrometry (PRM), ideally with isotope-labeled standards.
That final PRM step matters because it gives you an absolute, auditable number rather than a relative band intensity. Done well, the output can be expressed as a loading efficiency proxy such as "molecules-per-vesicle," linked to your particle count and recovery.
A practical encapsulation control set often includes:
| Control | What it tests | How to interpret |
|---|---|---|
| Protease only (no lysis) | Surface cargo removal | Signal should drop if cargo was surface-adhered |
| Protease + detergent | Total cargo accessibility | Signal should collapse (everything digested) |
| No protease + lysis | Total associated cargo | Defines upper bound (surface + internal) |
| RNase/DNase variants (if nucleic acid cargo) | External nucleic acids | Confirms nuclease-accessible material is removed |
In Vivo Biodistribution (BD) and PK Tracking Artifacts
If therapeutic EVs do not reach the intended target tissue in vivo, in vitro potency data alone may not be sufficient to support translational development decisions. But biodistribution is also one of the easiest places to generate convincing, wrong data.
The central risk is that you end up tracking the label, not the EV.
Mitigating Lipophilic Dye Artifacts
Lipophilic dyes such as PKH26, DiI, and DiR are commonly used because they are straightforward to apply.
One concrete example is the formation of non-EV fluorescent particles during labeling. A recent paper in Journal of Extracellular Vesicles reports this directly: Non-Specific Particle Formation During Extracellular Vesicle Labelling With the Lipophilic Membrane Dye PKH26 (PubMed). These particles can be indistinguishable from labeled EVs by size-based methods and can drive false localization in biodistribution readouts.
Beyond particle artifacts, dyes can dissociate in vivo and redistribute onto abundant circulating components. In practice, that often looks like exaggerated liver and spleen "EV uptake," when what you're actually seeing is dye behaving like a lipophilic tracer in a complex biofluid.
Robust programs reduce this risk with orthogonal strategies that track the EV itself, its engineered surface protein, or the therapeutic cargo.
| Tracking approach | What you're really measuring | Typical strengths | Common pitfalls |
|---|---|---|---|
| Lipophilic dyes (PKH/DiI/DiR) | Dye localization | Simple, low barrier | Dye aggregates; dye transfer; false positives |
| Genetic reporters (e.g., CD63-fusion) | Reporter-associated EV signal | Higher specificity; compatible with longitudinal imaging | Reporter may alter EV biology; needs stable engineering |
| Radiolabeling | Whole-particle label | Quantitative, sensitive | Label stability and chelation chemistry must be validated |
| Species-specific or targeted MS | Cargo or engineered protein peptides | Tracks biologically meaningful analytes | Requires assay development; tissue matrix effects |
The key is to design BD/PK so your readout answers the question you care about: "Did functional cargo reach the target tissue?" Not merely "Did a fluorescent signal accumulate somewhere?"
Surface Engineering and Potency Assay Alignment
Engineered EVs are moving fast: targeting ligands fused to membrane scaffolds (PTGFRN, Lamp2b, and others), immune-modulatory displays, and combinatorial surface designs. But surface engineering creates a new characterization problem: the engineered feature is often the mechanism of action.
If you can't prove the surface display is present, correctly oriented, and present at a controlled density, then potency data becomes difficult to interpret and comparability becomes fragile.
Verifying Surface Display and MoA-Linked Potency
Developers typically need to answer three "CMC-grade" questions:
- Is the targeting moiety on the EV surface, not just in the producer cell?
- Is it externally exposed with the correct topology (accessible to the target)?
- Is the display density stable lot-to-lot and across scale?
One practical approach is surface proteolytic shaving paired with high-resolution peptide mapping. Conceptually, you treat intact EVs with controlled proteolysis to sample exposed domains, then use LC-MS/MS to map peptides and quantify signature fragments. That gives you evidence for surface accessibility, and it can be paired with targeted PRM assays as a release-friendly readout.
This is also where potency assays need to stop being "generic uptake." A potency assay should be tied to your mechanism, for example:
- receptor engagement and downstream signaling
- target-cell internalization under competitive conditions
- functional cargo activity in the target tissue or relevant primary cells
If you're developing engineered EVs and need support across cargo loading, surface design, and characterization, exosome engineering can help align the engineering approach with assayable CQAs.
Navigating IND submission for a therapeutic EV requires phase-appropriate analytical evidence: deep identity and impurity profiling when you're defining CQAs, targeted quantitative assays when you're moving toward release, and orthogonal in vivo tracking when you're de-risking PK/BD. Creative Proteomics supports therapeutic EV development with mass spectrometry-based identity and impurity profiling, custom PRM assays for encapsulated cargo quantification, and EV-focused proteomics workflows designed to generate structured datasets for development and comparability decisions.
Frequently Asked Questions
Why is NTA insufficient for releasing therapeutic exosome batches?
NTA measures particle size and concentration based on light scattering, not EV identity. It cannot distinguish a loaded EV from an empty EV, a membrane fragment, or a similarly sized aggregate. For release and comparability, you typically need orthogonal identity and purity evidence (for example, marker confirmation plus proteomic fingerprinting and impurity profiling).
How can I prove my protein drug is actually encapsulated inside the EV?
Use a protease protection assay to remove surface-adhered cargo, then lyse the EVs and quantify the protected protein with a targeted assay such as PRM LC-MS/MS. The key deliverable is an absolute, auditable value that can be normalized to particle number, rather than a qualitative band or bulk signal.
Why do lipophilic dyes often produce false biodistribution data for exosomes?
Lipophilic dyes can form non-EV fluorescent particles and can also dissociate and redistribute in vivo, so the signal may reflect dye behavior rather than EV trafficking. Artifact risk is documented for PKH dyes, including in Non-Specific Particle Formation During Extracellular Vesicle Labelling With the Lipophilic Membrane Dye PKH26 (PubMed). To reduce false positives, use orthogonal tracking strategies that measure EV-associated reporters, radiolabel stability, or cargo-derived peptides.
What are the most important CQAs for therapeutic extracellular vesicles?
The most important CQAs are the ones that control patient risk and mechanism: identity (EV-associated markers and proteome fingerprint), purity (HCP and non-EV contaminants), cargo amount (encapsulated dose per vesicle), safety (procoagulant or immune-active proteins), and MoA-linked potency. The point is not to measure everything; it's to measure the few attributes that predict safety and function, with orthogonal methods.
How do I demonstrate batch comparability when scaling EV manufacturing?
Define a reference fingerprint (core EV proteome plus impurity signature), then show that process changes preserve the fingerprint while maintaining or improving impurity clearance. In practice, comparability packages often combine untargeted LC-MS/MS (to detect drift) with targeted panels (to monitor must-control impurities and engineered surface peptides).
How do I validate engineered exosome surface display?
Validate surface display with evidence of external accessibility and controlled density. Common strategies include antibody-based binding with appropriate controls, plus protease shaving and LC-MS/MS peptide mapping to confirm topology. For release, translate the mapping result into a targeted quantitative assay (e.g., PRM) that is stable across lots.
References
- Minimal information for studies of extracellular vesicles (MISEV2023): updated guidelines of the International Society for Extracellular Vesicles
- Mass-spectrometry-based molecular characterization of extracellular vesicles
- Circulating extracellular vesicles are endowed with tissue factor activity: focus on the procoagulant implications in COVID-19
- Non-Specific Particle Formation During Extracellular Vesicle Labelling With the Lipophilic Membrane Dye PKH26
* For Research Use Only. Not for use in diagnostic procedures.
