
Drug-induced mitochondrial toxicity can remain undetected in standard cell-viability assays and later emerge as hepatotoxicity, cardiotoxicity, or fatigue-related tolerability issues in vivo. The common failure mode is not the absence of mitochondrial assessment, but the use of early screens that fail to capture liabilities under bioenergetic demand.
What has changed in recent preclinical practice is the expectation of orthogonal evidence. Instead of treating mitochondrial liability as a single assay call, many teams now use a tiered pipeline: (1) sensitized functional screening to flag and rank compounds, (2) study design that separates primary mitotoxicity from downstream collapse, and (3) mechanistic resolution using subcellular multi-omics to generate actionable medicinal chemistry hypotheses and defensible Weight-of-Evidence (WoE) narratives.
Key takeaway: Endpoint cytotoxicity can miss mitochondrial liabilities. A tiered in vitro-to-omics workflow can identify drug-induced mitochondrial toxicity earlier and support more actionable mechanism-of-liability decisions.
The Preclinical Blind Spot: Why Standard Cytotoxicity Assays Miss Mitochondrial Liability
| Where standard screens fail | What you miss | What to add instead |
|---|---|---|
| Single endpoint after long incubation (MTT/CCK-8) | Early bioenergetic collapse, compensation, and adaptive stress | Real-time OCR/ECAR plus early Δ\Psi_m imaging |
| High-glucose culture in immortalized lines | Mitochondrial poisons masked by glycolytic ATP | Glucose→galactose sensitization to force OXPHOS reliance |
| One assay, one verdict | Ambiguous calls and late surprises | Tiered orthogonal evidence (functional + omics + kinetics) |
Late-stage attrition from organ toxicity is expensive for a simple reason: it often arrives after a compound has already been "validated" by routine in vitro screens. An MTT or CCK-8 readout can appear clean at doses that later associate with microvesicular steatosis, arrhythmogenic stress, or other organ-level safety concerns. For mitochondrial liabilities, the disconnect is structural. Many cytotoxicity assays are single endpoints measured after a long incubation, and they collapse a complex sequence of mitochondrial events into one final number.
A second, deeper issue is model metabolism. Many commonly used immortalized cell lines are cultured in high-glucose media and sit in a glycolysis-dominant energy state. That state can mask a surprisingly broad class of mitochondrial poisons: if the cell doesn't need oxidative phosphorylation (OXPHOS) to survive, OXPHOS inhibition won't look toxic until much later.
This phenomenon is often discussed through the Crabtree effect, where proliferative cells favor glycolysis even when oxygen is available. In that context, mitochondrial impairment can be "buffered" by glycolytic ATP, leading to false-negative mitochondrial toxicity screens. The practical implication is clear in classic work showing that switching from glucose to galactose increases sensitivity to mitochondrial toxicants in HepG2 models, because galactose forces greater reliance on OXPHOS for ATP production (see Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants). A detailed protocol perspective is also available in Assessment of mitochondrial toxicity in HepG2 cells cultured in high glucose- or galactose-containing media.
From roughly 2020 onward, the practical shift has been away from relying on endpoint viability alone and toward tiered workflows that combine functional phenotyping with mechanistic evidence. That shift aligns with broader regulatory and scientific expectations: if a signal is ambiguous, you strengthen your conclusion by adding independent, mechanistically coherent layers.
Tier 1: Real-Time Functional Screening and Sensitization Models
Tier 1 is about scale and speed. The goal is to rapidly identify and rank compounds with mitotoxic potential before a program is too committed. A good Tier 1 toolkit is deliberately phenotypic: it measures mitochondrial function directly (or sensitizes cells to require it), uses multiple readouts that fail for different reasons, and produces interpretable signatures.
The fastest way to increase confidence at this stage is to design Tier 1 readouts as a set rather than a single gate. A compound that causes (a) a glucose-to-galactose sensitization shift, (b) an OCR phenotype consistent with ETC inhibition or uncoupling, and (c) an acute Δ\Psi_m drop is qualitatively different from a compound that only drops viability at late timepoints.
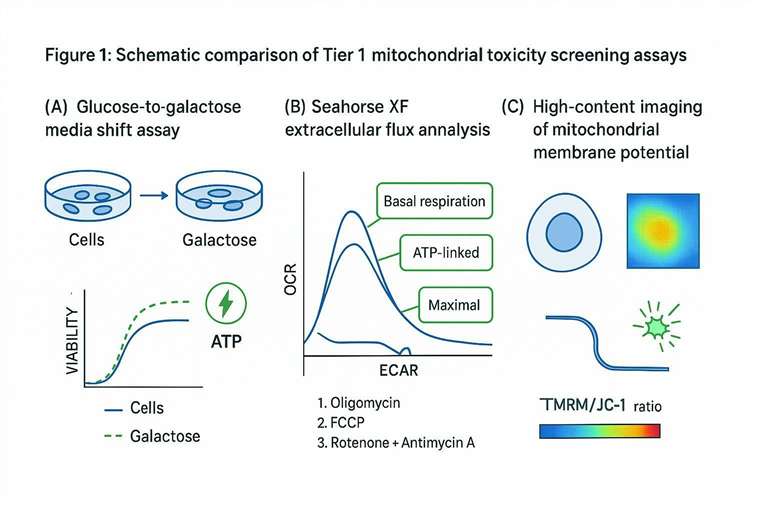
The Glucose-Galactose Media Shift Assay
The glucose-to-galactose shift is a simple intervention with outsized impact: you change the energy economics of the cell. Galactose yields little net ATP through glycolysis, so cells must lean harder on mitochondrial respiration to maintain ATP.
Mechanistically, this doesn't "prove" a compound is a mitochondrial toxicant. What it does is unmask dependence. If a compound looks tolerable in glucose media but becomes toxic in galactose, that differential suggests the cell is losing a mitochondrial function it can no longer compensate for.
A practical way to think about this assay is as a sensitized viability readout. You still measure viability or ATP, but you interpret the shift between conditions as the primary signal. Many labs apply empirical criteria for a "meaningful shift," and practical discussions often cite multi-fold IC_50 differences as a red-flag heuristic (see the practical overview in Mitochondrial Toxicity (Glu/Gal Assay)).
A few design details that matter more than they seem:
- Controls: include known ETC inhibitors (e.g., rotenone, antimycin A), ATP synthase inhibitors (oligomycin), and uncouplers (FCCP/CCCP) to confirm that the assay produces the expected response pattern.
- Normalization: interpret viability alongside cell number or protein normalization when possible; galactose can change growth rates.
- Exposure timing: acute mitochondrial poisons can show early ATP effects, but some liabilities are delayed and require longer exposure windows.
This assay is often the quickest way to reduce false negatives caused by glucose-fed, glycolysis-heavy culture conditions.
Extracellular Flux Analysis (Seahorse XF)
Extracellular flux analysis moves you from "did the cells die?" to "what happened to their energy system right now?" Seahorse XF instruments measure oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in live cells, giving you a real-time bioenergetic profile under metabolic pressure.
The workhorse format is the mitochondrial stress test, where sequential injections perturb OXPHOS in a controlled way:
- oligomycin (ATP synthase inhibition)
- FCCP (uncoupling to reveal maximal respiration)
- rotenone + antimycin A (block complexes I and III to define non-mitochondrial respiration)
From these traces you can compute interpretable parameters such as basal respiration, ATP-linked respiration, proton leak, maximal respiration, and spare respiratory capacity. Those parameters are useful because many mitochondrial liabilities don't present as complete OCR collapse. They present as reduced spare capacity or an abnormal response to uncoupling, which can be exactly what matters under in vivo demand.
Tier 1 Seahorse work is most powerful when it's paired with a clear interpretation question: Does the compound look like an ETC inhibitor, an uncoupler, or a more complex phenotype with glycolytic compensation? Adding ECAR helps because cells can compensate for mitochondrial inhibition by pushing glycolysis, masking the "severity" of OCR effects if you only look at viability.
If your program needs a validated, end-to-end functional readout package (OCR/ECAR plus complementary mitochondrial integrity measures), a practical starting point is Mitochondrial function analysis.
High-Content Imaging of Mitochondrial Membrane Potential (ΔΨm)
Mitochondrial membrane potential (Δ\Psi_m) is one of the earliest functional indicators of mitochondrial distress. Potentiometric dyes such as TMRM/TMRE or ratio dyes like JC-1 can detect acute depolarization events that may occur before overt cell death.
High-content imaging changes the value of these dyes because you gain two things: kinetics and context. Instead of a single well-average fluorescence value, you can measure distributions, identify subpopulations, and link Δ\Psi_m changes to morphology (fragmentation, swelling) and oxidative stress proxies.
A few practical cautions prevent this readout from becoming noisy:
- Controls are non-negotiable: FCCP/CCCP as depolarization controls; consider ROS controls if you're reading MitoSOX or related dyes.
- Dye concentration matters: quenching and dye-induced perturbation are common failure modes for TMRM/TMRE.
- Interpretation must be paired: Δ\Psi_m loss alone doesn't tell you whether the primary insult is ETC inhibition, uncoupling, or permeability transition.
For high-throughput imaging-oriented mitochondrial phenotyping workflows, Mitochondrial phenotype analysis using high-content imaging is the relevant internal resource.
Tier 2: Distinguishing Primary Mitotoxicity from Secondary Cytotoxic Effects
Tier 2 is where drug-induced mitochondrial toxicity becomes a decision you can defend. The goal is not to generate more data. It's to show, with timing and dose discipline, that mitochondrial dysfunction is an initiating event rather than an end-stage artifact.
Tier 1 identifies a potential mitochondrial liability; Tier 2 defines whether that liability reflects primary mitochondrial dysfunction or secondary cytotoxic collapse.
The core mandate in investigative toxicology is attribution: is the compound directly poisoning the mitochondrion (primary mitotoxicity), or is the mitochondrial dysfunction a downstream artifact of global stress, membrane damage, or the late stages of apoptosis/necrosis?
This distinction matters because the remediation strategies are different. Primary mitotoxicity can sometimes be mitigated through medicinal chemistry once you know the target interaction or the chemical liability. Secondary mitochondrial collapse usually means you should investigate the upstream mechanism or off-target cytotoxicity pathway.
Tier 2 is largely about experimental design. The two most powerful levers are:
- Sub-cytotoxic dosing: select concentrations that preserve overall viability while still producing measurable mitochondrial perturbation.
- Time-course kinetics: sample early enough that you're still inside the "pre-lethal window," before generalized proteome degradation, calcium overload, and membrane rupture dominate.
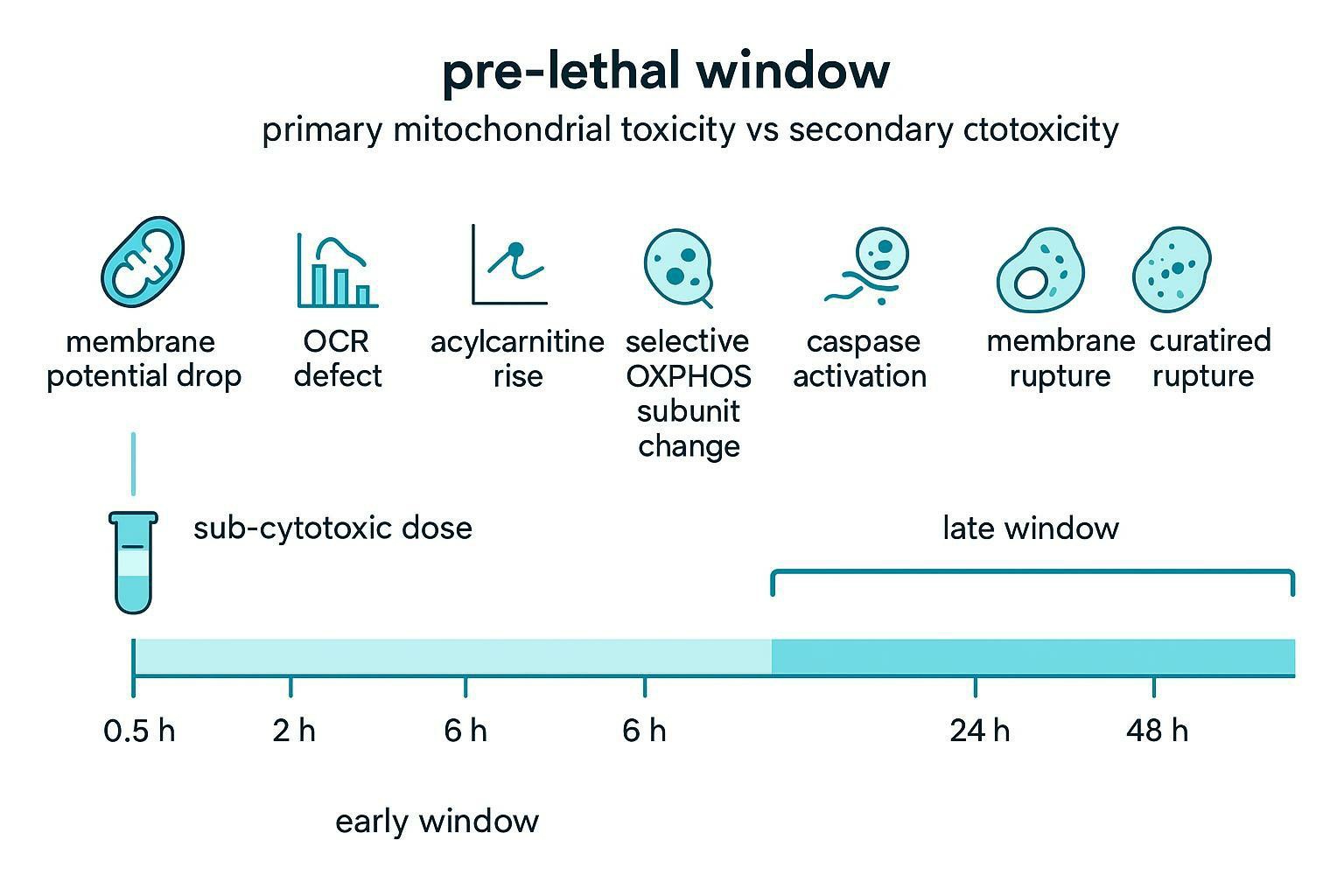
A useful way to make this operational is to define a chronology of signals. Mitochondria often show a structured sequence when they are directly targeted: early Δ\Psi_m changes, early OCR defects, specific metabolite signatures, and selective pathway-level proteomic changes. By contrast, many forms of general cytotoxicity produce broad, non-specific degradation across compartments at roughly the same time.
The table below is not a law of nature, but it is a practical pattern that helps teams avoid mistaking late-stage debris for mechanism.
| Signal timing / pattern | What you observe | Interpretation bias | How to de-confound |
|---|---|---|---|
| Early (minutes to hours) Δ\Psi_m drop at sub-lethal dose | TMRM/TMRE decrease or JC-1 ratio drop before viability loss | Supports primary mitochondrial involvement | Pair with OCR/ECAR and confirm cell number normalization |
| Early OCR defect with abnormal stress-test response | Reduced spare capacity / maximal respiration; phenotype consistent with ETC inhibition or uncoupling | Supports primary mitochondrial dysfunction | Include positive controls; compare glucose vs galactose |
| Early metabolite shifts in mitochondrial pathways | Acylcarnitine accumulation; TCA intermediate bottlenecks | Supports pathway-specific energetic impairment | Sample in pre-lethal window; compare to general stress controls |
| Selective proteomic signature | Coordinated change in OXPHOS assembly factors, mitoribosome proteins, mitophagy network | Supports mechanistic specificity | Use subcellular enrichment; replicate across timepoints |
| Late (24–48 h) generalized collapse | Global proteome degradation; membrane rupture; apoptosis markers dominate | Often secondary | Move sampling earlier; use lower doses |
Operationally, Tier 2 success depends on prioritizing the cleanest mechanistic window rather than the largest apparent effect size.
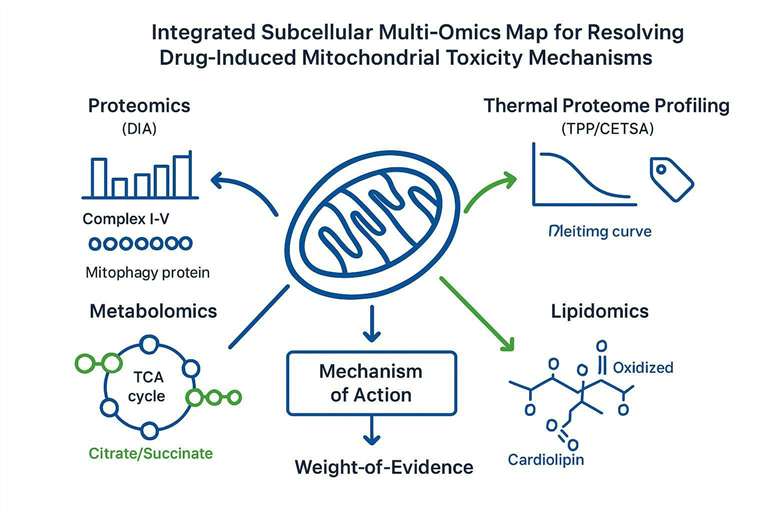
Subcellular Multi-Omics for Molecular Target Resolution
Once you have a credible mitochondrial liability signal, the next question is no longer "is mitochondria involved?" It is "what exact molecular event is failing, and can we change it?"
This is where subcellular multi-omics is at its best. A mitochondrion is not one target. It's a network: membranes, electron transport chain complexes, transporters, translation machinery, turnover pathways, and lipid structures that hold the whole system together.
A mechanistic workflow typically benefits from two design principles:
- Subcellular enrichment: improve sensitivity and specificity by enriching mitochondria or mitochondrial membranes before deep MS profiling.
- Orthogonal causality: use at least one method that can support target engagement (TPP/CETSA) rather than relying on downstream expression changes alone.
Proteomics, Thermal Proteome Profiling (TPP), and Translation Deficiencies
Deep quantitative proteomics can detect the coordinated disruption patterns that are hard to see in single-marker assays. DIA proteomics, in particular, is often chosen for its reproducibility and lower missingness across complex samples.
In drug-induced mitochondrial toxicity, common proteomic questions include:
- Are respiratory complexes I–V selectively down-regulated, disassembled, or missing key subunits?
- Do assembly factors or supercomplex-associated proteins show coordinated shifts?
- Is there evidence of mitophagy activation or organelle quality-control engagement?
A useful conceptual reference for MS-based mitochondrial proteomics is Proteomics as a Tool for the Study of Mitochondrial…. For workflows specifically aligned with organelle-level mitochondrial profiling, Mitochondrial proteomics services is the relevant internal service link.
Where proteomics becomes mechanistically decisive is when you combine it with target engagement strategies.
Thermal Proteome Profiling (TPP) and CETSA-MS approaches quantify drug-associated changes in protein thermal stability across the proteome. The logic is straightforward: if a protein's stability shifts reproducibly in the presence of a compound, that can point toward direct binding or a tightly coupled complex-level perturbation.
A representative CETSA-MS study showing proteome-scale thermal profiling for compound interaction mapping is available as CETSA MS Profiling for a Comparative Assessment of FDA-Approved Anticancer Drugs. The key practical benefit for mitochondrial toxicology is that TPP/CETSA can turn "mitochondrial dysfunction" into a shortlist of candidate interacting proteins or complexes.
Finally, mitochondrial liabilities often intersect with mitochondrial translation and mitonuclear coordination. Because OXPHOS complexes contain both mitochondrial-encoded and nuclear-encoded subunits, a translational blockade can produce an imbalanced stoichiometry: nuclear-encoded components may remain stable while mitochondrial-encoded subunits drop, or vice versa, creating assembly stress. Quantifying these ratios and their time dependence can provide strong mechanistic clues even when the compound doesn't obviously "target mitochondria" in a classical sense.
Metabolomics and Lipidome Hallmarks of Membrane Injury
Proteomics tells you what pathways and complexes are changing. Metabolomics tells you what those changes are doing to flux.
In mitochondrial toxicity, high-resolution metabolomics often reveals pathway-level bottlenecks that map cleanly to mechanistic hypotheses:
- TCA cycle bottlenecks: accumulation of upstream organic acids consistent with enzymatic inhibition or redox constraints.
- Fatty acid oxidation stress: accumulation of short-, medium-, or long-chain acylcarnitines consistent with incomplete oxidation.
For a practical overview of acylcarnitines as mitochondrial biomarkers, see L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Disease. When your mechanistic question is specifically mitochondrial metabolism, Mitochondrial metabolomics analysis services is the most relevant internal link.
Lipidomics becomes particularly important when you suspect the inner mitochondrial membrane (IMM) is the primary injury site. The IMM is not just a barrier. It's a functional platform for OXPHOS, and its lipid composition is tightly linked to complex stability.
A hallmark lipid in this context is cardiolipin, which is enriched in the IMM and has well-studied roles in respiration and apoptosis signaling. Oxidation and remodeling of cardiolipin can precede, and contribute to, cell-death pathway activation. A strong mechanistic review is Cytochrome c/cardiolipin relations in mitochondria: a kiss of death, and classic mechanistic work includes Mechanisms of cardiolipin oxidation by cytochrome c and Cytochrome c-promoted cardiolipin oxidation generates singlet molecular oxygen and induces cell death.
A compact way to operationalize multi-omics interpretation is to map each omics layer to a "hallmark" and a next experiment.
| Omics layer | Hallmark signal | What it suggests | A practical next step |
|---|---|---|---|
| Proteomics (DIA) | Coordinated depletion/disassembly of complexes I–V | ETC complex destabilization or assembly defect | Add complexome profiling or targeted validation of complex subunits |
| TPP/CETSA-MS | Thermal stability shift of a mitochondrial protein/complex member | Candidate direct interaction or tight pathway coupling | Validate with orthogonal engagement (dose response, lysate vs intact cells) |
| Metabolomics | Acylcarnitine accumulation + TCA bottleneck pattern | FAO/TCA redox constraint; energetic failure under demand | Pair with OCR/ECAR; test substrate-rescue logic |
| Lipidomics | Cardiolipin remodeling/oxidation signature | IMM structural injury and oxidative stress linkage | Evaluate ROS kinetics, cytochrome c release markers, membrane integrity |
Data Integration and the Regulatory Weight-of-Evidence (WoE) Framework
This is where many programs tighten their conclusion around drug-induced mitochondrial toxicity: the story shifts from assay readouts to a coherent chain of causality supported by independent evidence layers.
Multi-omics data is only as useful as the story you can defend with it. In a regulatory or governance context, that usually means moving from "we saw changes" to "we have a coherent causal narrative supported by independent evidence."
A practical WoE package for drug-induced mitochondrial toxicity often integrates:
- Tier 1: sensitized viability shifts (Glu/Gal), OCR/ECAR phenotypes, Δ\Psi_m imaging signatures
- Tier 2: pre-lethal window time-course evidence showing mitochondrial readouts change before global cytotoxicity
- Tier 3: mechanistic resolution demonstrating pathway specificity and, when possible, target engagement (TPP/CETSA)
- Biomarkers: mass-spectrometry-verified panels (proteins, metabolites, cardiolipin species) that can be tracked across models
Even when mitochondrial toxicity is not treated as a standalone endpoint, regulators and reviewers expect integrated reasoning for complex safety signals. For liver-injury risk assessment, the FDA's guidance Drug-Induced Liver Injury: Premarketing Clinical Evaluation and the EMA's Reflection paper on non-clinical evaluation of drug-induced liver injury (DILI) illustrate how safety decisions are grounded in integrated interpretation rather than a single assay. More broadly, the nonclinical safety framework is laid out in ICH M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization.
In practice, a WoE narrative becomes much easier to defend when you can point to at least one causal anchor: a target engagement signal, a pathway bottleneck that maps to enzyme logic, or a structural membrane hallmark (such as cardiolipin oxidation) that precedes downstream death pathways.
Building a Defensible Mitochondrial Toxicity Weight-of-Evidence Package
If your team needs to evaluate a mitochondrial liability under governance or regulatory scrutiny, the key challenge is not simply running additional assays. It is connecting functional phenotypes to subcellular mechanisms and interpretable biomarkers. Mitochondrial analysis services can support integrated workflows that link Tier 1 functional screening, including OCR/ECAR, glucose–galactose sensitization, and ΔΨm imaging, with deeper subcellular MS layers such as DIA proteomics, CETSA/TPP-style target engagement strategies, metabolomics, and cardiolipin-focused lipidomics. This integrated approach can help move a suspected liability toward a defensible Weight-of-Evidence package.
Frequently Asked Questions
How do I know if my compound is causing primary mitochondrial toxicity or just secondary cytotoxicity?
Primary mitochondrial toxicity is most likely when mitochondrial readouts change at sub-cytotoxic doses before general viability loss, and when the same phenotype appears across orthogonal assay types (for example, OCR defects plus early Δ\Psi_m loss). Secondary mitochondrial dysfunction is more likely when mitochondrial changes only appear at high, broadly cytotoxic doses and track tightly with late-stage cell death markers.
What does a glucose–galactose shift actually tell me?
A glucose–galactose shift tells you whether your cells can compensate for mitochondrial impairment by relying on glycolysis. If toxicity increases meaningfully in galactose, it suggests the compound compromises mitochondrial ATP production or respiratory capacity in a way that becomes lethal when OXPHOS is required.
What does Seahorse OCR/ECAR add beyond an ATP or viability assay?
Seahorse OCR/ECAR adds a functional phenotype that can separate mitochondrial inhibition from glycolytic compensation. In practice, parameters like spare respiratory capacity and the response to oligomycin/FCCP can reveal liability signatures long before an endpoint viability assay turns negative.
Should I use JC-1 or TMRM/TMRE for mitochondrial membrane potential in screening?
TMRM/TMRE is often preferred for quantitative workflows because the readout can be more controllable, but either dye can work if you validate loading conditions and include depolarization controls (such as FCCP/CCCP). The key is to interpret Δ\Psi_m changes alongside cell count normalization and at least one independent mitochondrial functional readout.
How can I avoid false positives from "general stress" in mitochondrial toxicity assays?
Use a pre-defined pre-lethal window time course and focus on sub-cytotoxic doses, then check whether the earliest changes are mitochondria-specific rather than global. If the first major signals are generalized membrane damage, broad proteome collapse, or late apoptosis markers, you're likely looking at secondary effects.
What is the regulatory Weight-of-Evidence (WoE) framework, and how does multi-omics fit into it?
A WoE framework is an integrated approach that combines independent data layers to support a safety conclusion when any single assay is ambiguous. Multi-omics fits by providing mechanistic specificity: a Tier 1 phenotype (like an OCR defect) becomes more defensible when it aligns with a proteomic signature (complex disruption), a metabolomic bottleneck (TCA/FAO stress), or a lipidomic hallmark (cardiolipin injury).
What is Thermal Proteome Profiling (TPP), and how does it help salvage a candidate with mitotoxicity signals?
TPP (and related CETSA-MS approaches) measures drug-associated shifts in protein thermal stability that can point to target engagement or tight pathway coupling. If you can identify a specific mitochondrial protein or complex perturbed by the compound, medicinal chemistry can often prioritize structural changes that reduce that interaction while preserving the desired on-target activity.
Can multi-omics distinguish primary mitochondrial toxicity from secondary effects?
Often yes, if sampling is done before global cell death. Primary mitochondrial mechanisms tend to produce structured, pathway-coherent omics signatures (for example, selective OXPHOS assembly disruption or cardiolipin oxidation) rather than the broad, non-specific degradation patterns that dominate late-stage cytotoxicity.
Which assays are most informative for early lead optimization decisions?
A practical early set is a sensitized screen (glucose–galactose shift), a functional bioenergetics phenotype (OCR/ECAR stress test), and one orthogonal integrity marker (Δ\Psi_m imaging). That combination balances throughput with mechanistic interpretability.
Do I need isolated mitochondria to run a credible mechanistic investigation?
Not always, but enrichment can improve interpretability when you move into proteomics, lipidomics, or complex assembly questions. In many workflows, intact-cell phenotyping defines the liability, and subcellular enrichment strengthens the mechanistic readout by reducing background from non-mitochondrial compartments.
References
- Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants
- Assessment of mitochondrial toxicity in HepG2 cells cultured in high glucose- or galactose-containing media
- Mechanisms of cardiolipin oxidation by cytochrome c: relevance to proapoptotic properties of the protein
- Cytochrome c-promoted cardiolipin oxidation generates singlet molecular oxygen and induces cell death
- Cytochrome c/cardiolipin relations in mitochondria: a kiss of death
* For Research Use Only. Not for use in diagnostic procedures.
