CORE SERVICE
Global Ubiquitinome Profiling — Site-Level Identification and Quantification of Protein Ubiquitination
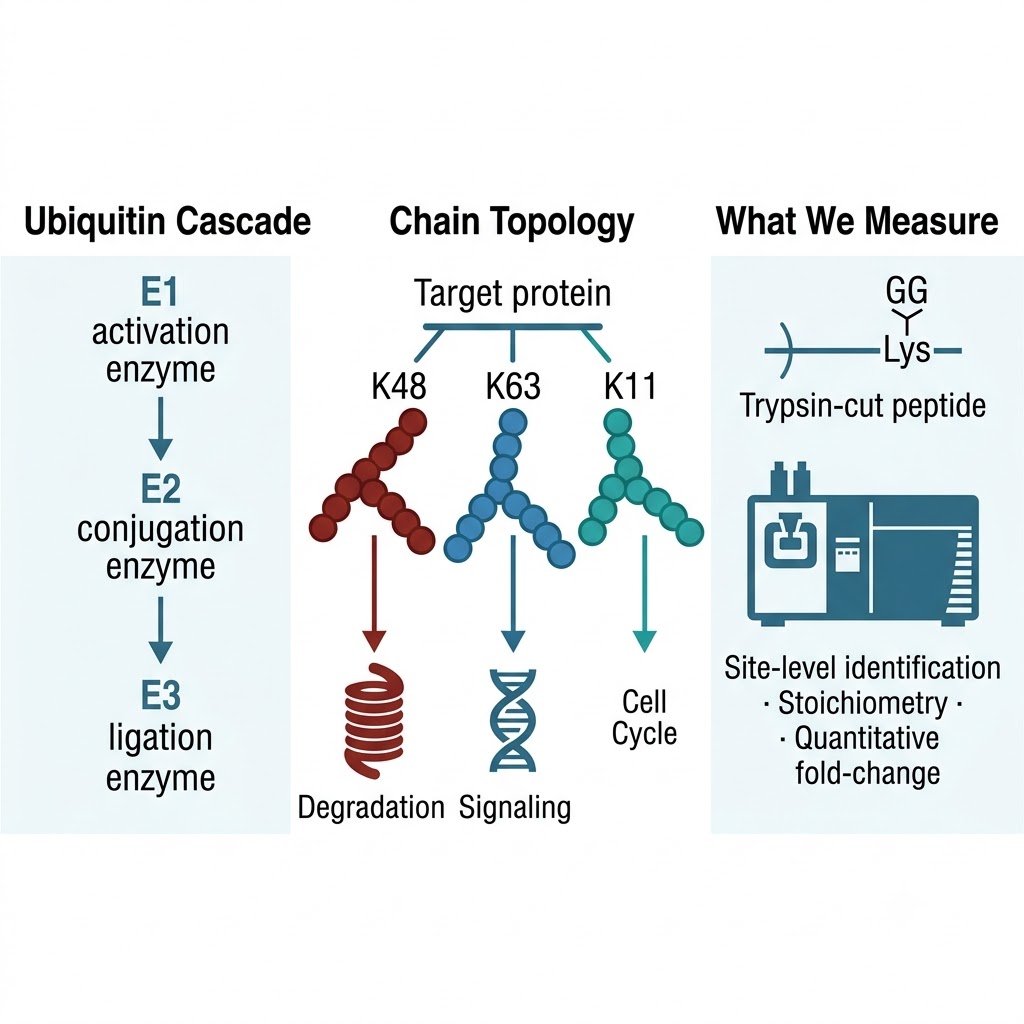
Protein ubiquitination is the master regulatory PTM of the ubiquitin-proteasome system (UPS) — controlling protein half-life, signal transduction, DNA repair, immune response, and virtually every other major cellular process. Unlike phosphorylation, which can be studied with generic kinase inhibitors, ubiquitination is mediated by a three-enzyme cascade (E1 → E2 → E3 ligase) in which the E3 confers substrate specificity. Identifying which proteins are ubiquitinated, on which specific lysine residues, at what stoichiometry, and how these events change in response to perturbations — drugs, gene knockouts, disease states — requires mass spectrometry-based ubiquitinome profiling at proteome scale.
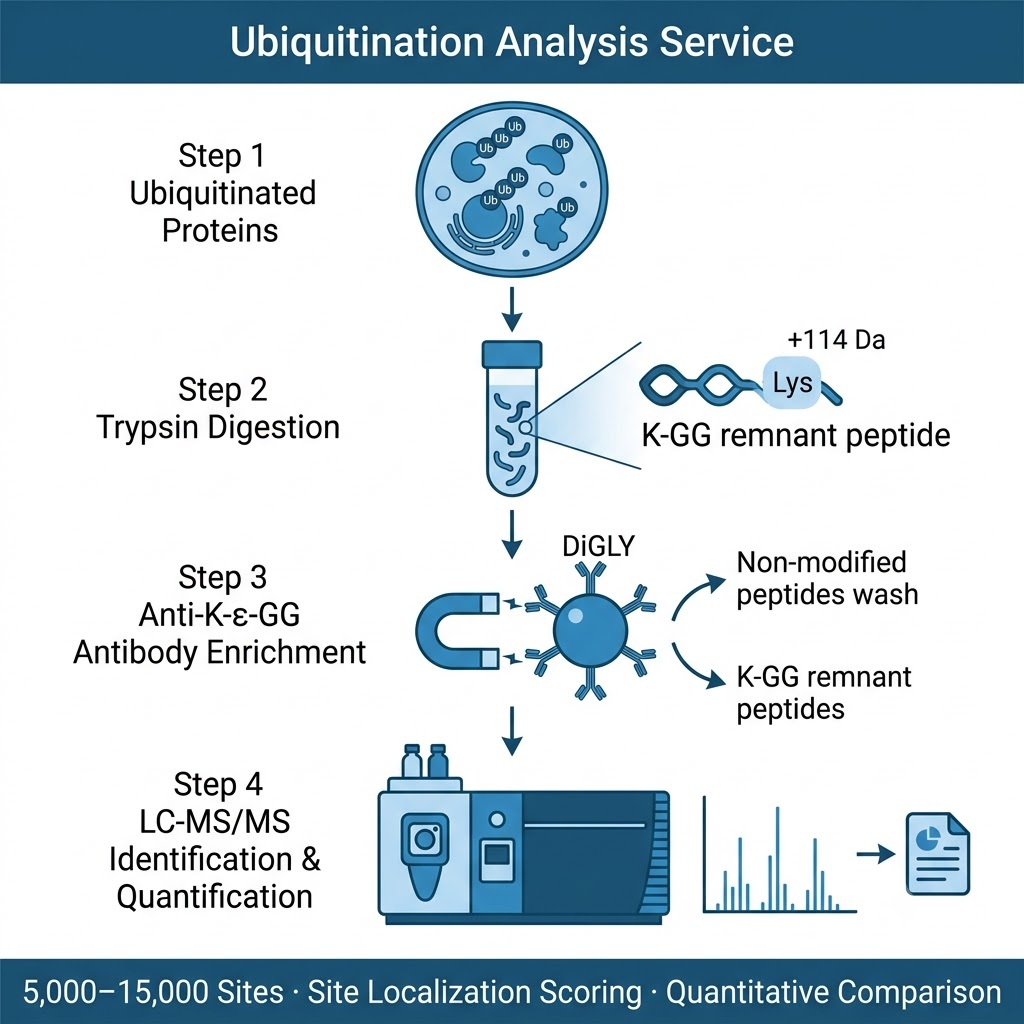
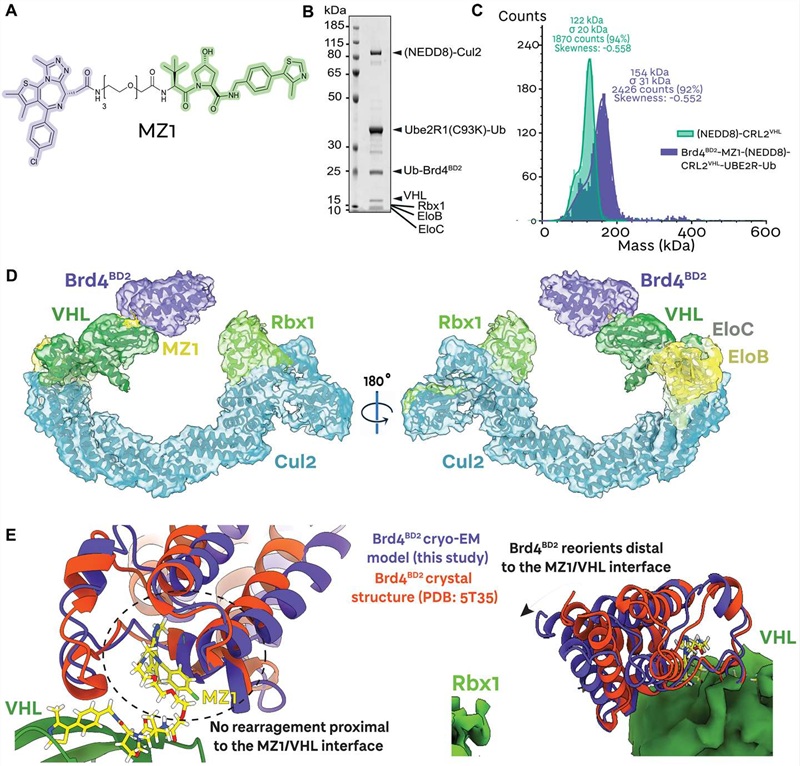
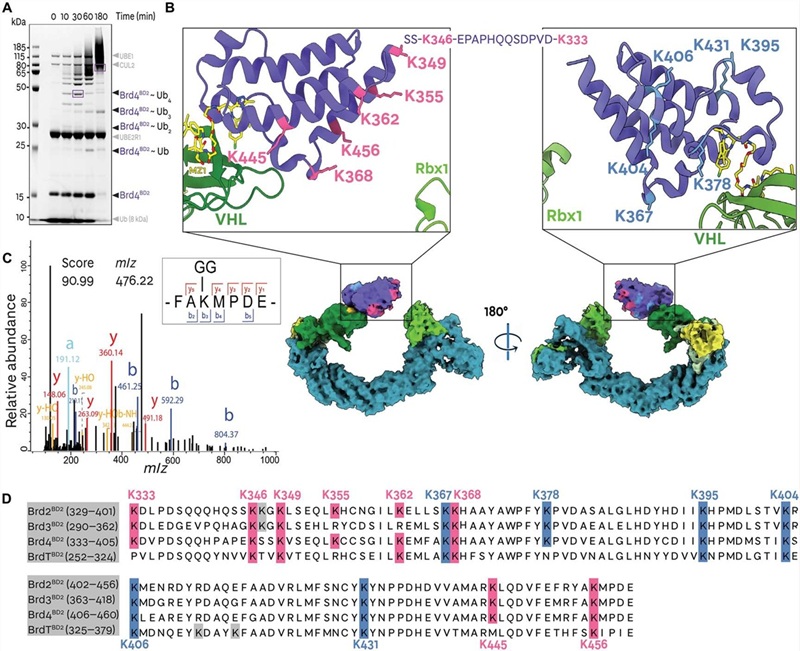
Our Ubiquitination Analysis Service uses the diGLY antibody immunoaffinity enrichment approach: after trypsin digestion, ubiquitinated proteins generate characteristic K-ε-Gly-Gly (diGLY) remnant-modified peptides that are specifically captured by a validated anti-diGLY monoclonal antibody (PTMScan K-ε-GG kit, Cell Signaling Technology), then analyzed by high-resolution LC-MS/MS. This approach identifies endogenous ubiquitination events without requiring expression of tagged ubiquitin, covers the full ubiquitinome in a single experiment, and can be coupled with quantitative strategies — SILAC, label-free, or TMT — to deliver fold-change and statistical significance data for every regulated site between conditions.
- Deep ubiquitinome coverage: Starting from ≥10 mg total protein per sample, our standard workflow identifies 5,000–15,000 unique ubiquitination sites across 2,000–5,000 proteins per experiment, with site localization probability scores ≥0.75 reported for all confident assignments. For PROTAC/degrader studies requiring focused analysis of a single target protein, lower input and shorter timelines are available.
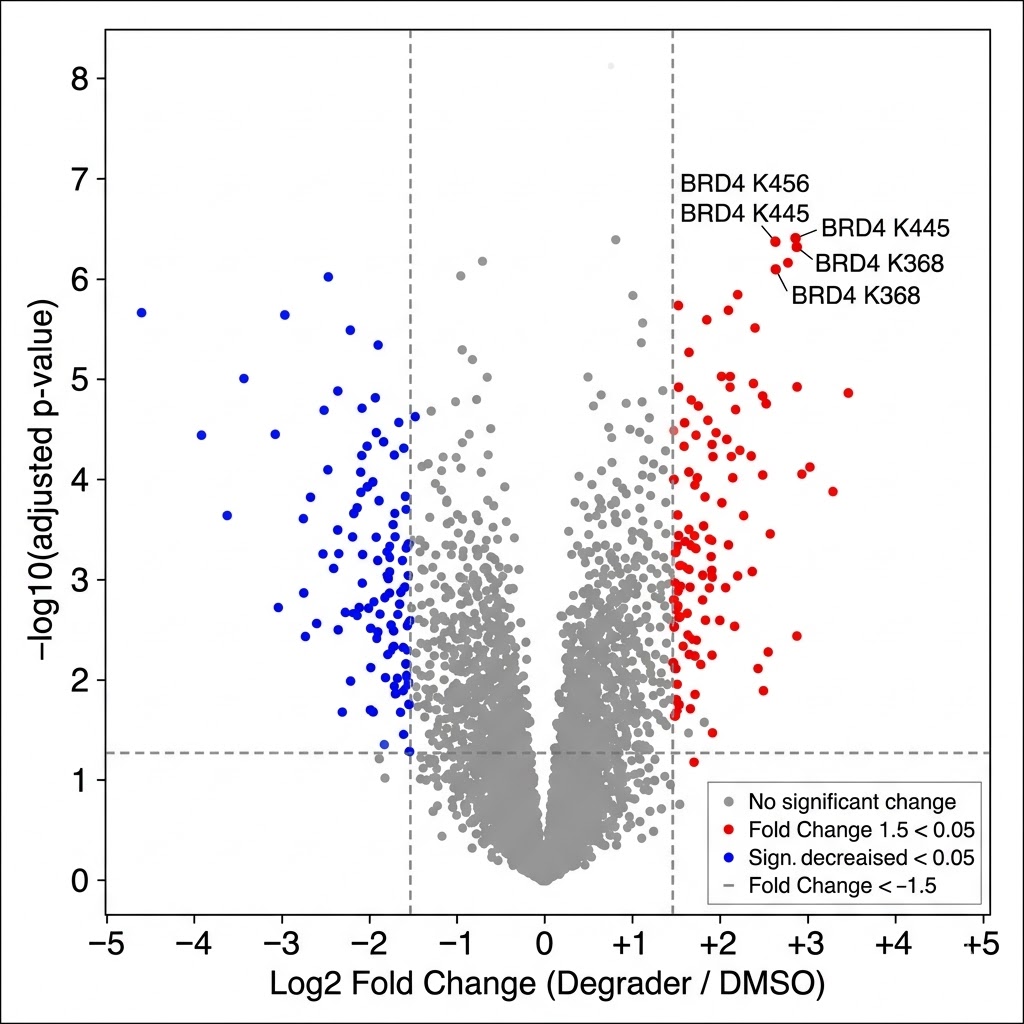
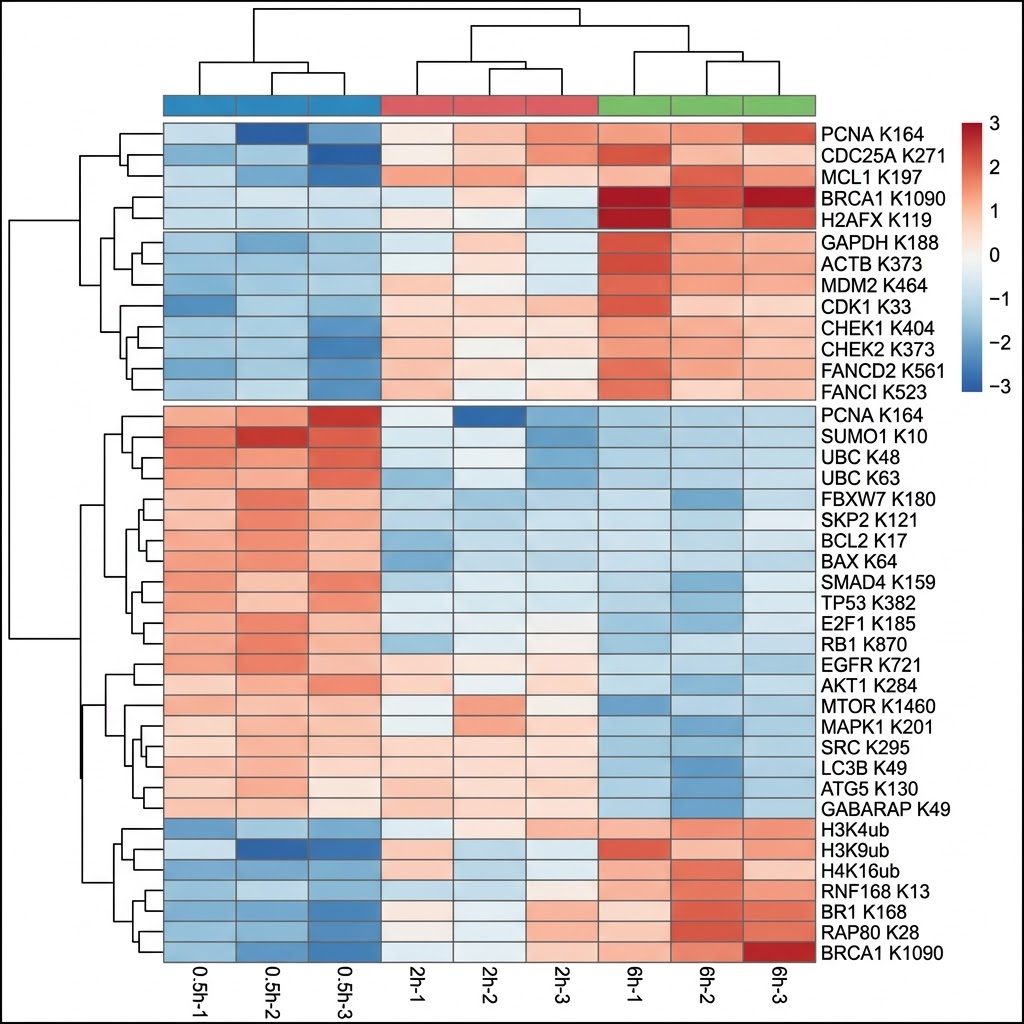
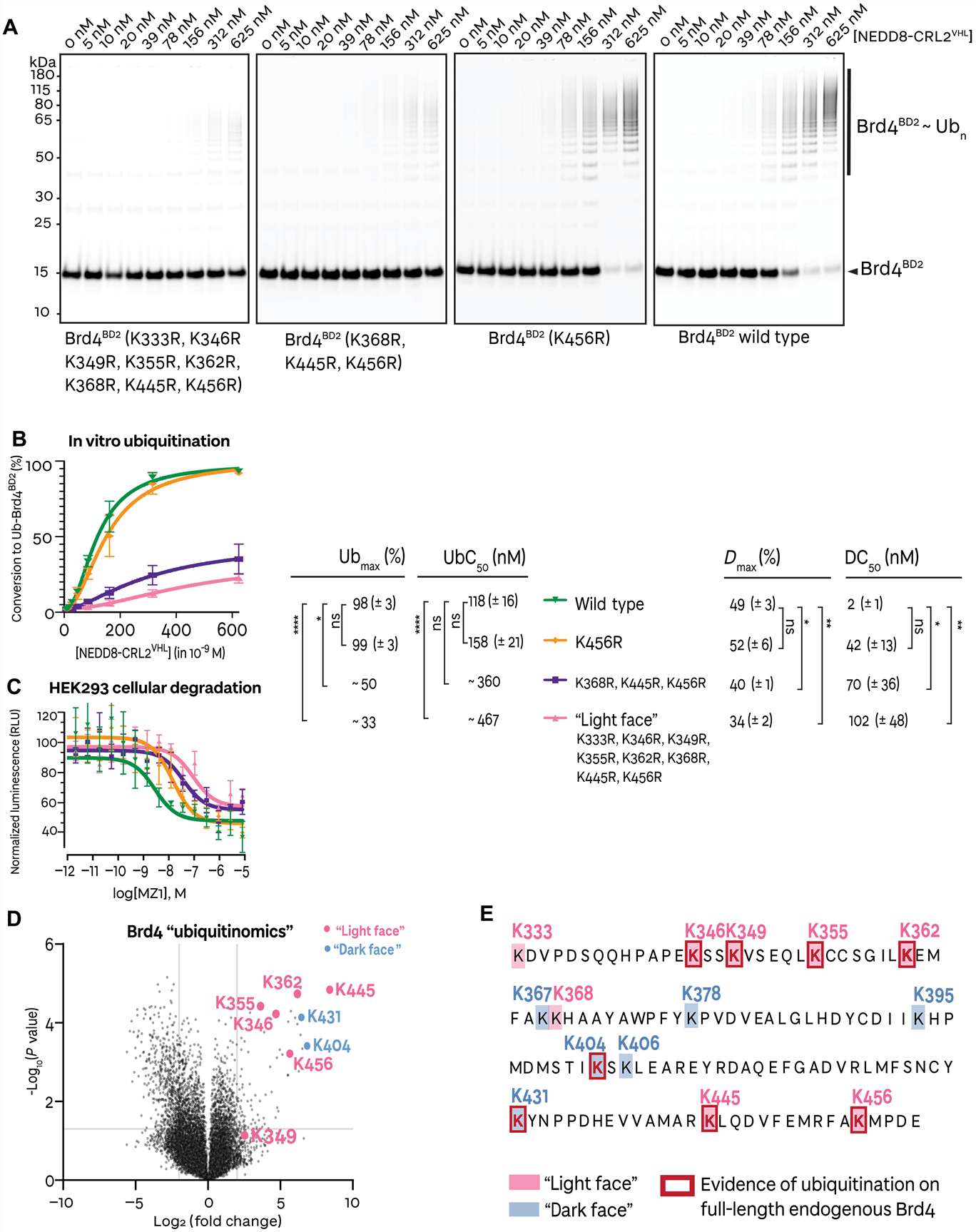
- Quantitative comparison across conditions: We apply SILAC pulse-labeling, label-free (MaxLFQ), or TMT multiplexing to generate site-level fold-change data between treated and control cells — identifying which ubiquitination events are gained, lost, or unchanged in response to proteasome inhibition, E3 ligase modulation, or small-molecule degrader treatment.
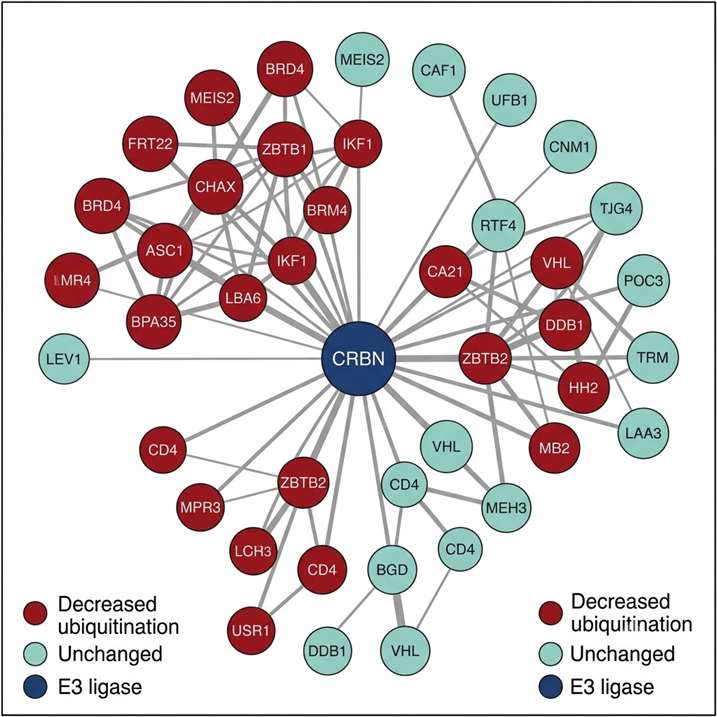
- Integrated UPS pathway annotation: Identified and regulated sites are cross-referenced against PhosphoSitePlus, UbiNet, and human protein interaction databases to annotate known E3 ligase-substrate relationships, ubiquitin chain topology (K48 vs. K63 linkage context), and downstream pathway connections — converting a site list into an interpretable UPS regulatory map.