CORE SERVICE
Full-Spectrum PTM & Signaling Proteomics — from Site Identification to Quantitative Pathway Analysis
Post-translational modifications are the molecular switches that control protein activity, localization, stability, and interaction at every level of cellular signaling. A single phosphorylation event can activate or silence an entire kinase cascade; ubiquitination determines whether a protein is degraded or re-routed; glycosylation governs cell-surface recognition and secretory trafficking; acetylation at histones reconfigures chromatin state and gene programs. To study these mechanisms with the resolution, quantitative precision, and throughput demanded by modern drug discovery and mechanistic research, mass spectrometry-based PTM proteomics is the definitive analytical approach — and the quality of the enrichment chemistry, the instrument platform, and the data analysis pipeline determines everything about what you can and cannot detect.
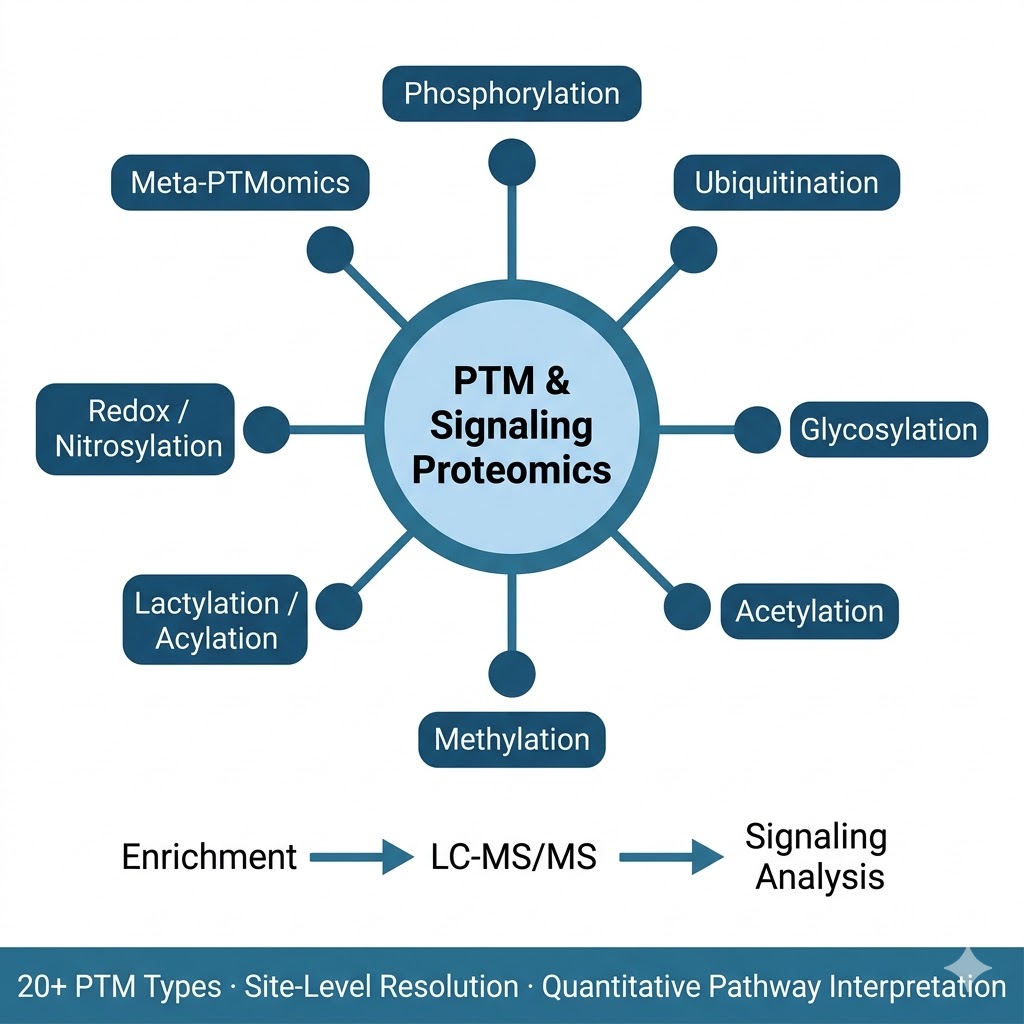
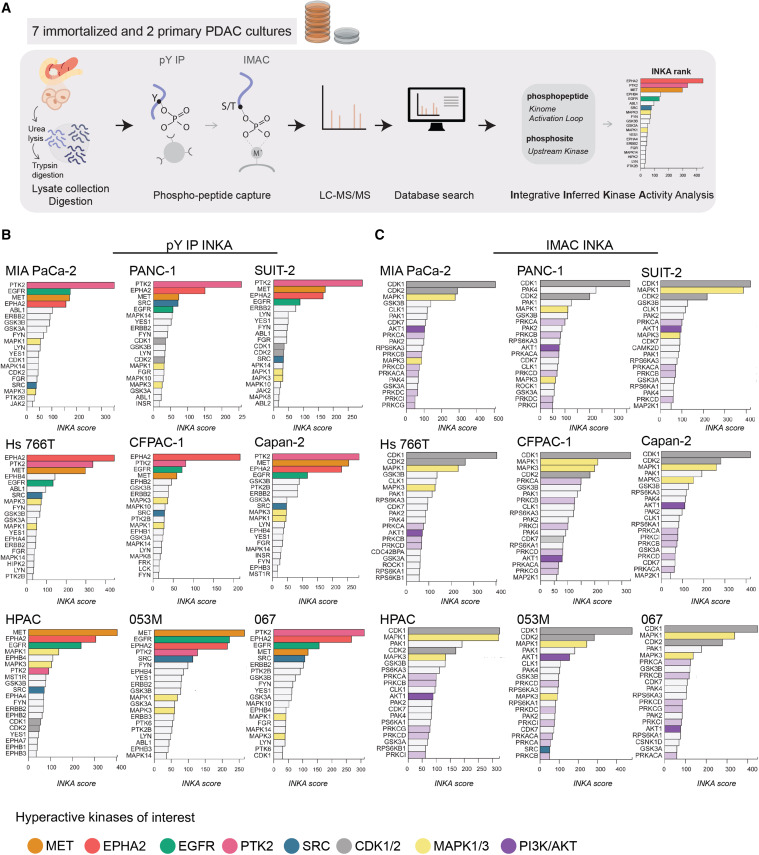
We provide the broadest PTM proteomics service portfolio available from a single CRO, covering more than 20 modification types across qualitative identification, site mapping, quantitative profiling, and integrative multi-PTM analysis. Our workflows combine modification-specific enrichment (IMAC/TiO2 for phospho, lectin affinity or HILIC for glyco, diGLY immunoprecipitation for ubiquitin remnants, antibody-based enrichment for acetyl and acyl marks) with high-resolution mass spectrometry on the Bruker timsTOF Pro, Thermo Orbitrap Fusion Lumos, and Q Exactive HF-X, and a dedicated PTM bioinformatics pipeline covering site localization scoring, stoichiometry calculation, kinase-substrate network inference, and pathway-level signaling analysis.
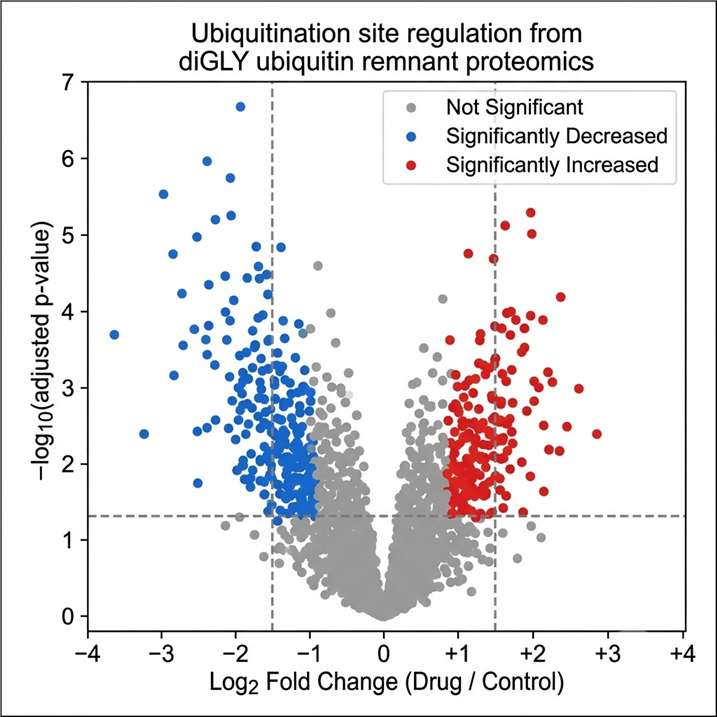
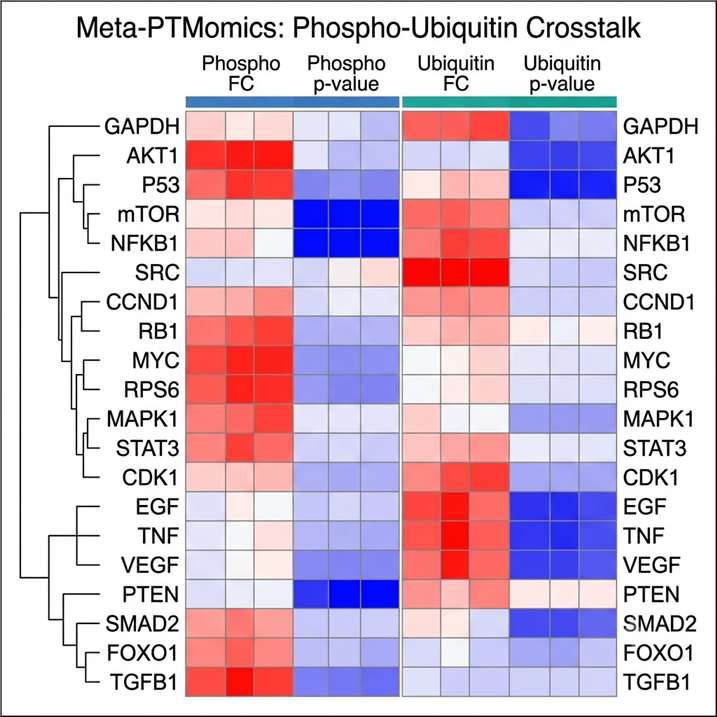
- Site-level resolution across 20+ PTM types: We cover the full range of biologically relevant modifications from the most studied (phosphorylation, N- and O-glycosylation, ubiquitination/diGLY) to the emerging (lactylation, crotonylation, succinylation, SUMOylation, nitrosylation, lipidation, disulfide bond mapping, and Meta-PTMomics integrative analysis), each with dedicated enrichment and detection protocols.
- Quantitative PTM analysis: Every profiling service can be extended to quantitative mode using label-free, TMT, or SILAC strategies, providing modification-site-level abundance ratios between conditions, stoichiometry estimates, and differential regulation statistics — not just presence/absence information.
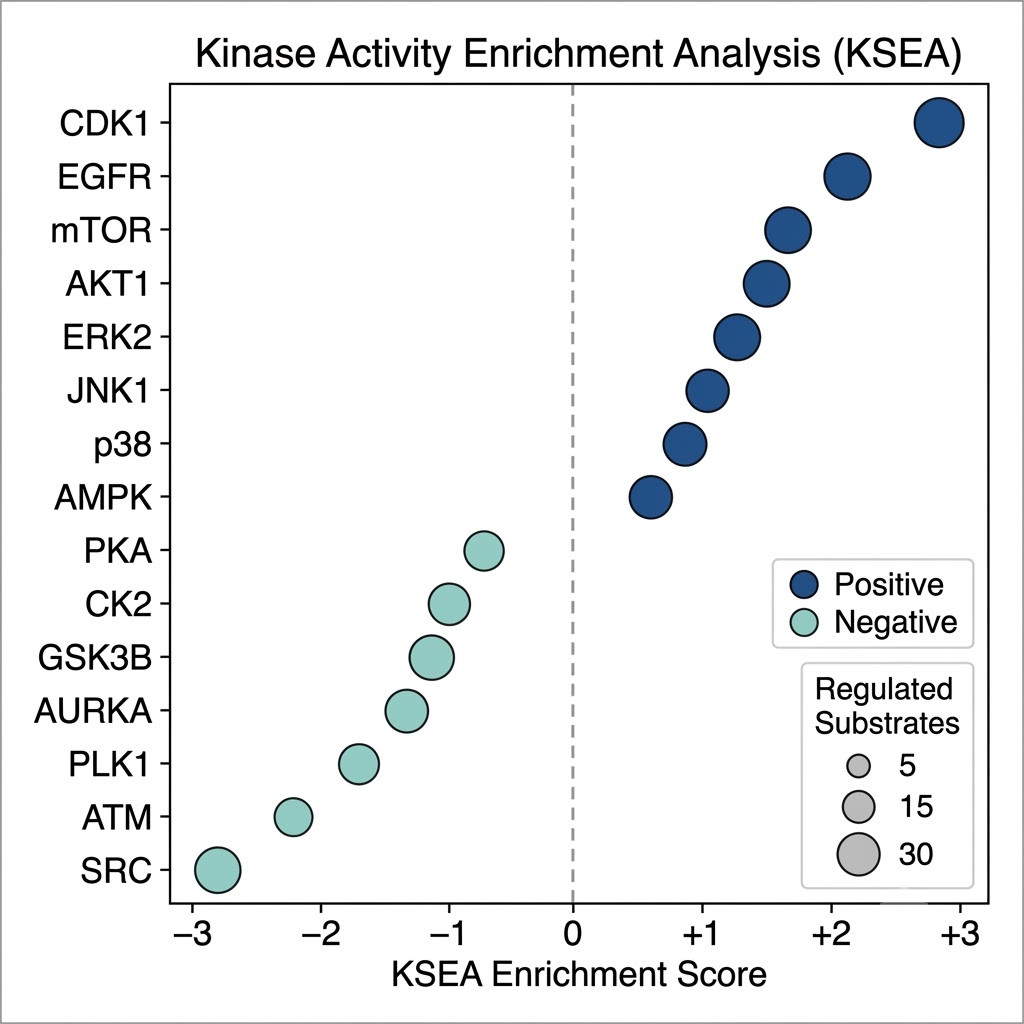
- Signaling pathway context: PTM data without functional context is a list of sites. Our bioinformatics pipeline connects identified and regulated modification sites to kinase-substrate databases (PhosphoSitePlus, NetworKIN), E3 ligase-substrate networks, glycan structure databases, and KEGG/Reactome signaling pathways, converting a raw modification inventory into an interpretable map of active signaling.