Integrating LiP-MS With Other Proteomics Solutions
Integrating with Thermal Proteome Profiling (TPP)
TPP measures how the overall stability of a protein responds to environmental factors or small molecules. Together, these methods reveal not only that a protein is affected, but also how it responds at a structural and stability level, giving a more complete picture of its functional state.
Integrating with Targeted Proteomics Services
After LiP-MS identifies structural changes across the proteome, targeted methods can validate and quantify these changes in specific proteins with high precision. This is particularly valuable when studying low-abundance proteins, membrane proteins, or proteins with complex modifications.
Integrated into Drug Discovery and Translational Proteomics
LiP-MS can also be integrated into Drug Discovery and Translational Proteomics workflow. Structural insights from LiP-MS reveal where compounds bind and how they affect protein conformation. When used alongside quantitative proteomics tools like DIA, researchers can track not only structural changes but also overall protein abundance, giving a multi-dimensional view of how molecules interact with cellular networks.
Applications of LiP-MS in Proteomics and Drug Discovery
Protein Structural Dynamics and Conformation Studies
LiP-MS enables high-resolution detection of conformational changes, providing insights into protein folding, post-translational modifications, and environment-induced structural alterations.
Mapping Protein-Protein and Protein-Small Molecule Interactions
LiP-MS identifies accessible regions affected by protein-protein and protein-small molecule interactions, allowing the study of protein networks and ligand binding. It is essential for understanding complex regulatory mechanisms in cellular pathways.
Drug Target Identification and Mechanistic Studies
By revealing drug-induced structural changes, LiP-MS assists in mapping compound binding sites and validating potential targets. Researchers can prioritize targets based on conformational impact, aiding mechanistic studies and structure-based research.
Biomarker Discovery in Disease Research
LiP-MS can uncover novel structural biomarkers, such as amyloid-to-soluble protein ratios in neurodegenerative disorders, providing insights into pathological mechanisms and protein dysfunction.
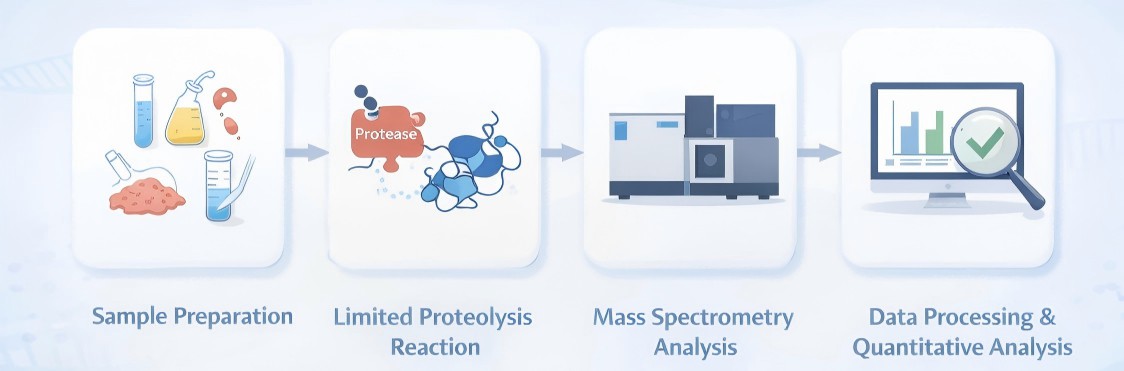
Creative Proteomics' LiP-MS Service Workflow
- Sample Preparation: Samples can include cells, tissues, protein solutions, serum, and cerebrospinal fluid. Proteins are extracted under non-denaturing conditions to preserve native conformations.
- Limited Proteolysis Reaction: A broad-spectrum or specific protease is applied to partially digest proteins. TProteolysis is stopped through denaturation, and proteins are subsequently digested with trypsin.
- Mass Spectrometry Analysis: Peptide fragments are separated by LC and analyzed by MS. LiP-MS supports multiple acquisition strategies. Quantitative analysis is typically performed in label-free mode or using stable isotope-labeled standards for absolute quantification.
- Data Processing and Quantitative Analysis: Advanced bioinformatics tools are employed to map peptide fragments to protein sequences, quantify changes, and identify statistically significant structural alterations.