CORE SERVICE
Full-Spectrum Discovery Proteomics — from Protein ID to Deep Quantification
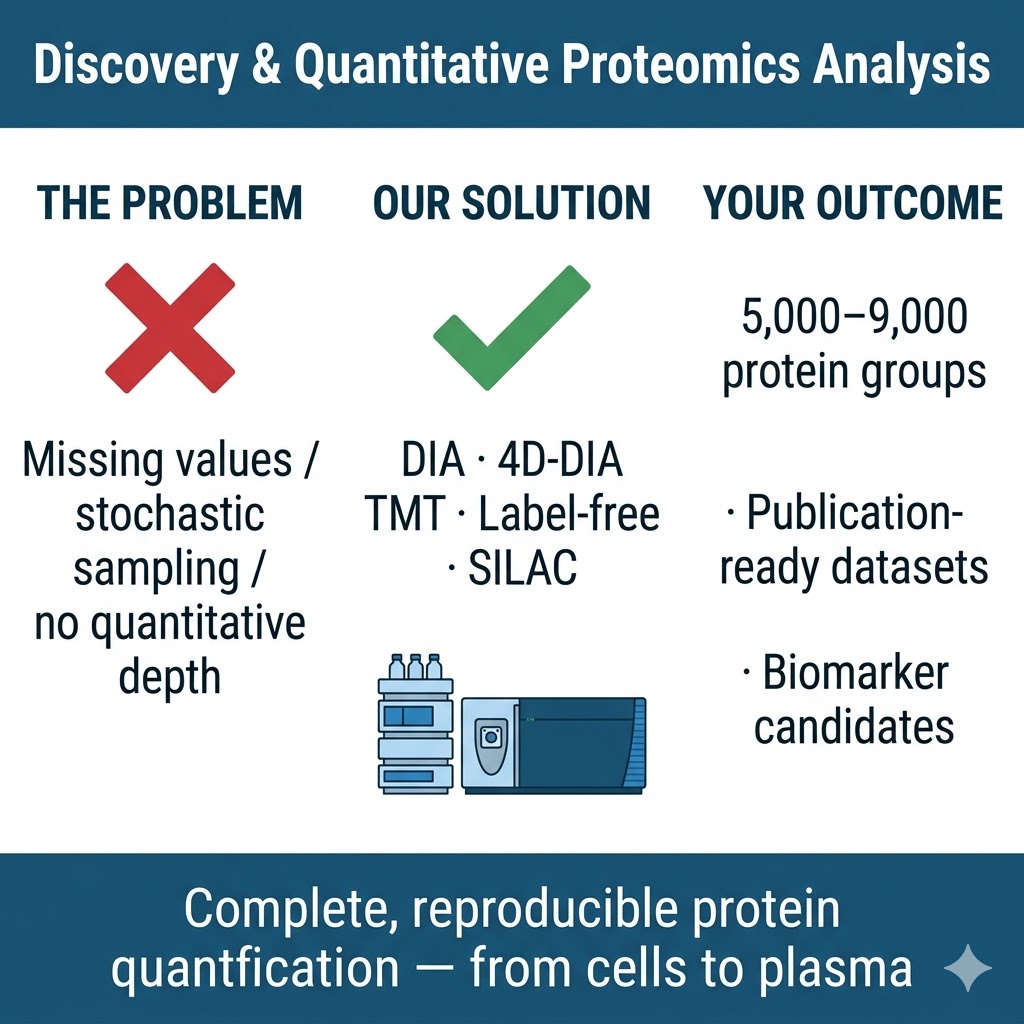
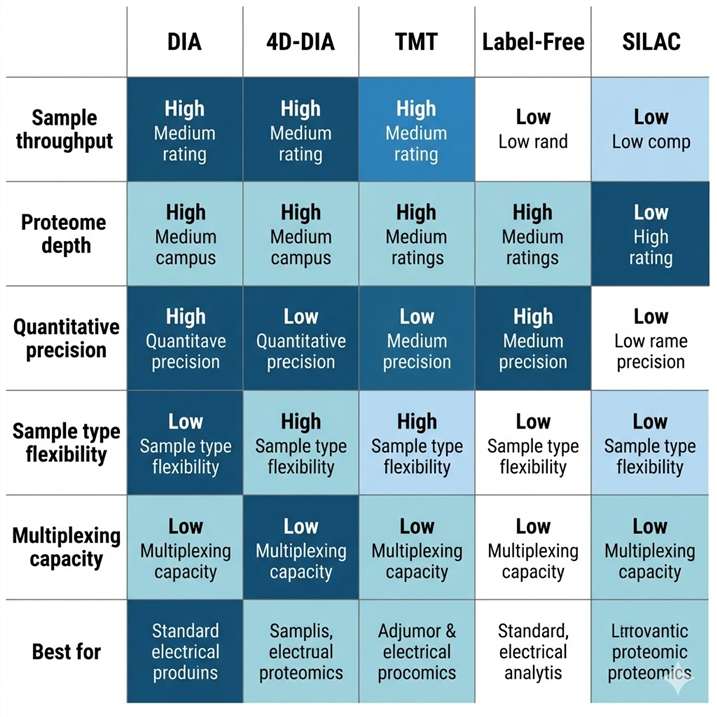
Discovery and quantitative proteomics form the foundation of every protein-level research program. Whether you need an unbiased snapshot of how the proteome shifts between conditions, or a deep, reproducible quantification dataset to support a biomarker discovery cohort, the right workflow choice determines what you can confidently measure, compare, and publish. We operate across the full range of established and next-generation discovery proteomics platforms — DIA, 4D-DIA (diaPASEF on timsTOF HT), label-free, 4D label-free, TMT, iTRAQ, and SILAC — all on high-resolution instruments including the Bruker timsTOF Pro, Thermo Q Exactive series, and Orbitrap Fusion Lumos, with standardized QC and publication-grade bioinformatics integrated into every project.
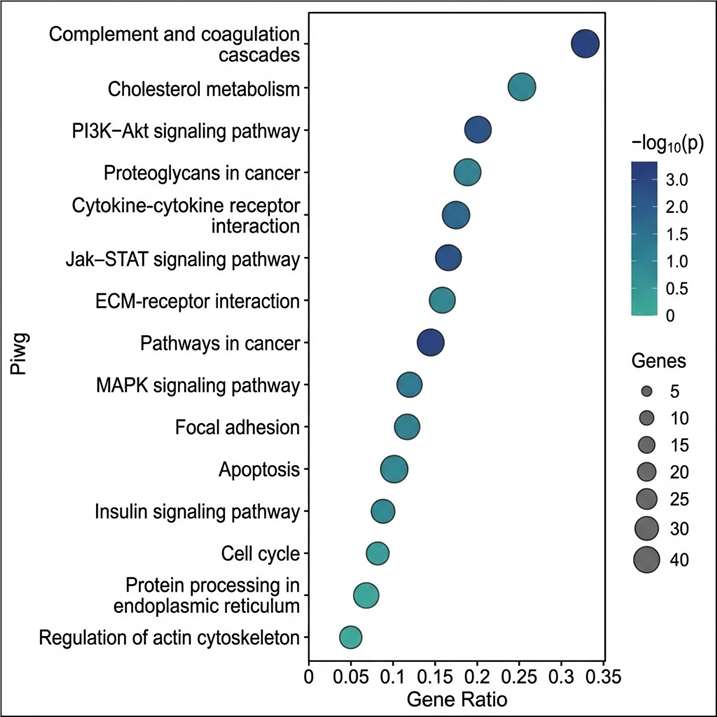
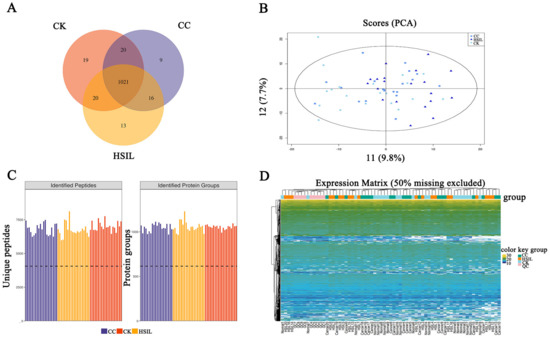
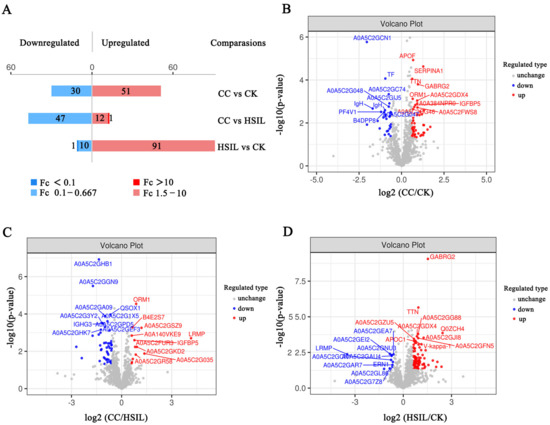
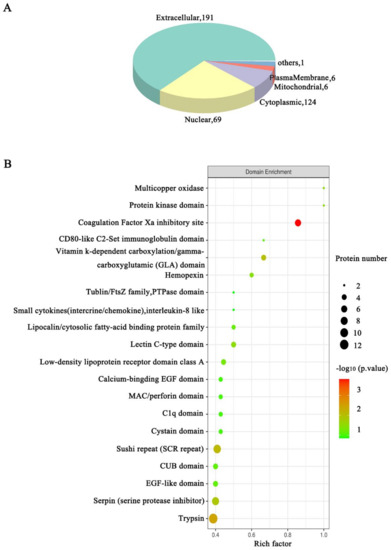
- Deep proteome coverage: DIA and 4D-DIA workflows routinely identify and quantify thousands of proteins per sample, with low missing-value rates even across large cohorts; ion mobility separation in 4D workflows adds a fourth dimension that reduces spectral interference and improves identification confidence for complex matrices such as plasma, FFPE tissue, and exosomes.
- Flexible quantification strategies: We match the workflow to your experimental design — label-free or 4D label-free for large or heterogeneous sample sets, TMT (up to 16-plex) or iTRAQ for high-throughput multiplexed comparisons, SILAC for cell-culture-based pulse-chase or interaction studies — so your quantitative data is optimized for your study, not the other way around.
- End-to-end project delivery: From sample intake QC through LC-MS/MS acquisition, database search, differential expression analysis, pathway enrichment, and final report with methods text, we deliver a comprehensive data package ready to support manuscript submission without additional processing.