CORE SERVICE
CSF Proteomics: Your Proximal Window into Brain Pathophysiology
Cerebrospinal fluid bathes the brain and spinal cord, carrying proteins secreted, shed, and released by neurons, glia, and the choroid plexus. Unlike plasma — where brain-derived proteins are diluted into a systemic protein pool orders of magnitude larger — CSF sits in direct contact with the central nervous system. A protein changing in the brain parenchyma changes in CSF. This makes CSF the most information-rich biofluid for studying neurodegenerative diseases, neuroinflammatory conditions, and CNS drug target engagement.
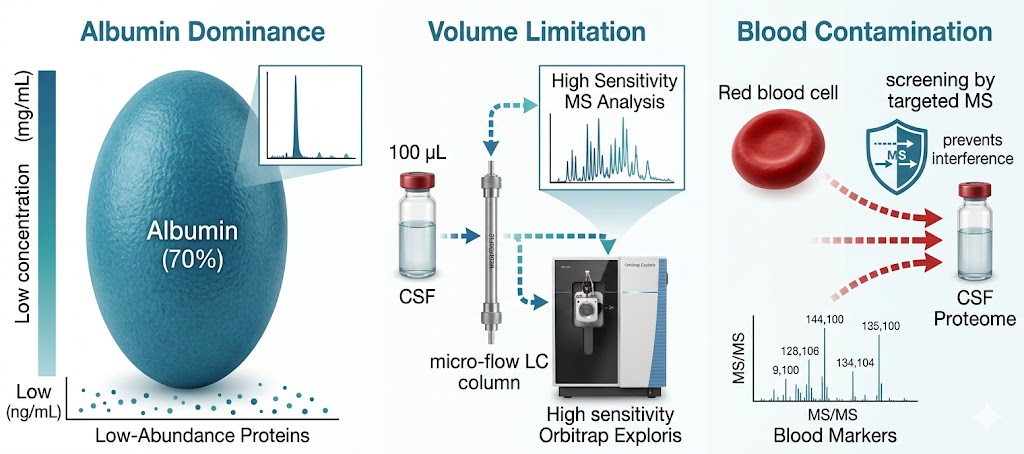
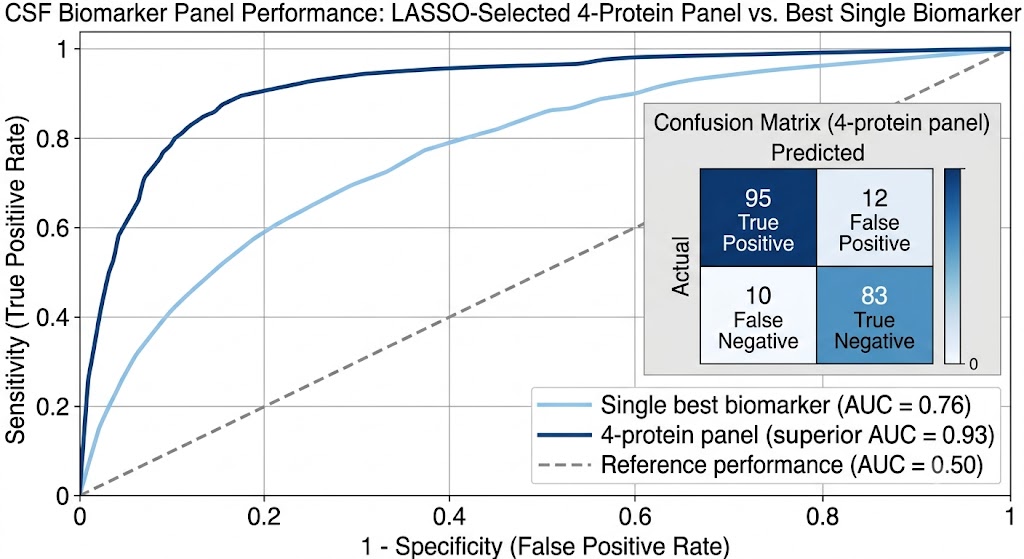
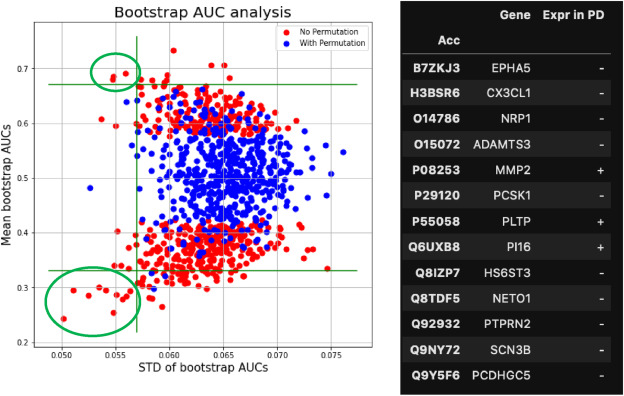
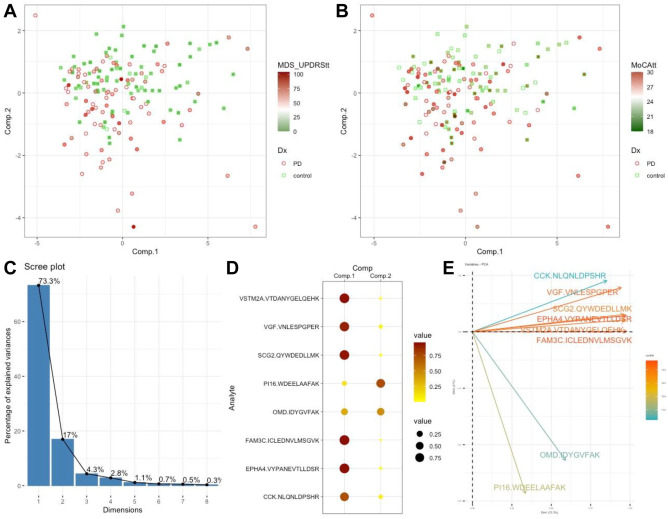
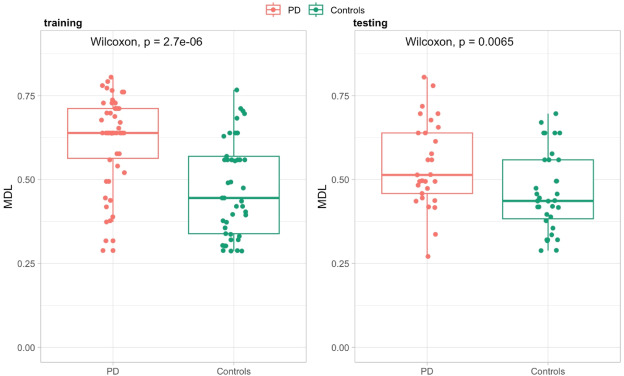
But CSF proteomics is harder than plasma proteomics. We treat CSF as a specialty matrix — not just another sample type on a general proteomics page. Our workflows are optimized for the specific analytical demands of cerebrospinal fluid, deploying immunoaffinity depletion, nanoparticle-based enrichment, and micro-flow LC gradients tuned for limited input volumes. From a typical 100–500 µL CSF sample, we routinely quantify 800–2,000+ protein groups.
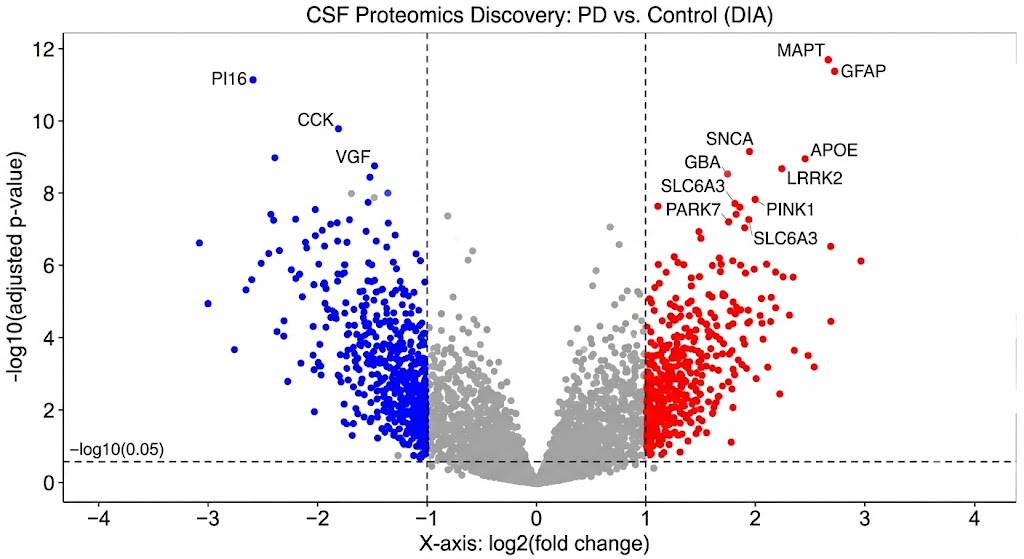
- Depth: DIA acquisition captures a complete digital record of every detectable CSF peptide — no stochastic precursor selection, no run-to-run gaps
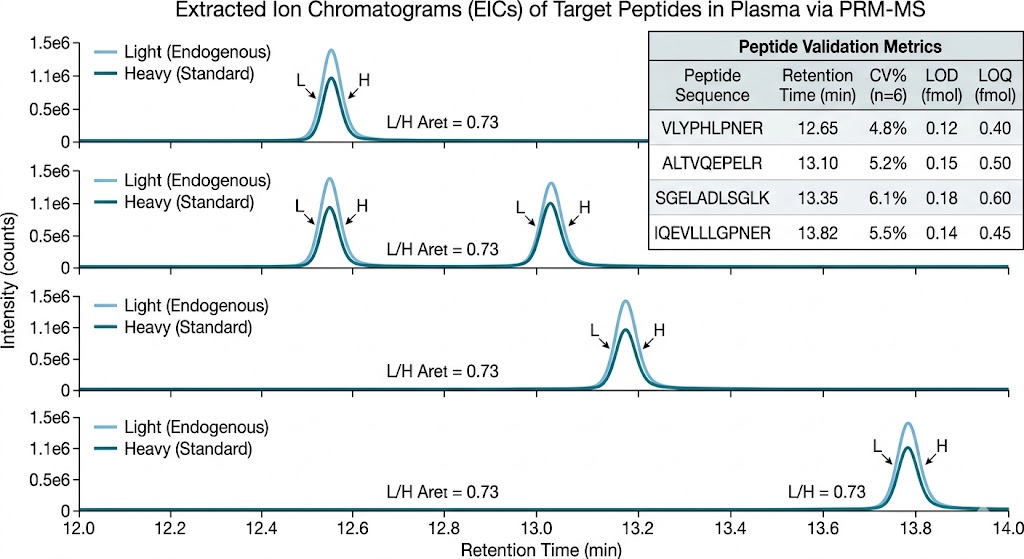
- Integration: The entire discovery-to-validation pipeline runs under one roof — from unbiased DIA discovery through PRM-targeted verification with heavy isotope-labeled standards to independent cohort validation
- Neuro-Expertise: We work with CSF from human lumbar puncture, ventricular drain, mouse cisterna magna, rat, and non-human primate collections, and we know the matrix-specific handling requirements for each