CORE SERVICE
Comprehensive Immunopeptidomics for T-Cell Immunotherapy Research
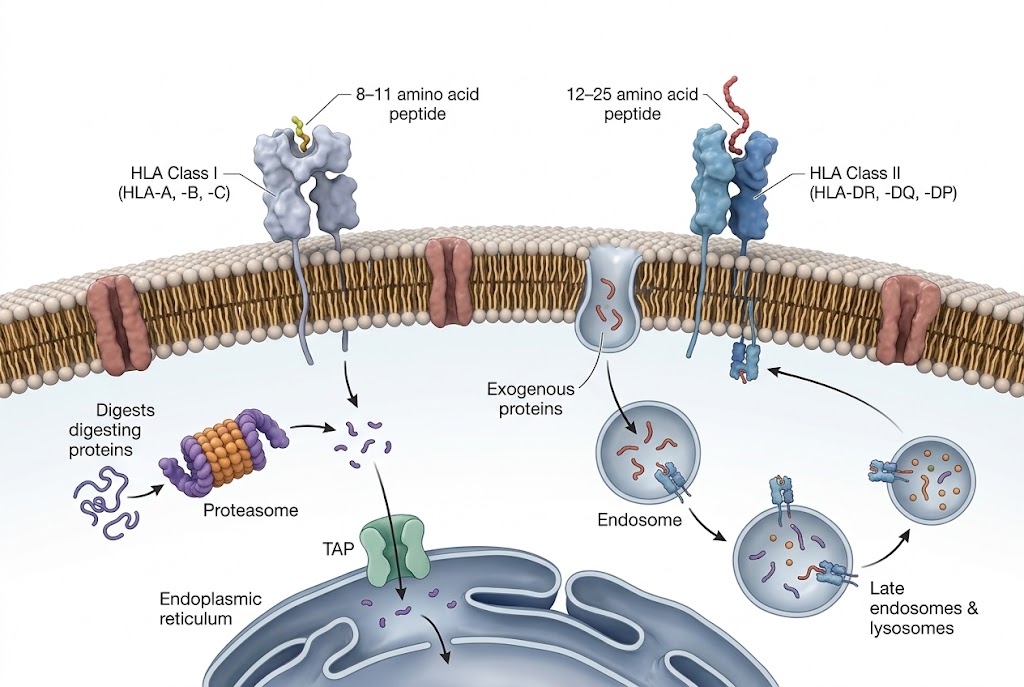
The peptides presented by major histocompatibility complex (MHC) molecules on the cell surface constitute the immune system's window into the cellular proteome. These HLA-bound peptide repertoires — the immunopeptidome — are the molecular basis for T-cell recognition in infection, autoimmunity, and cancer. Identifying which peptides are presented in a given biological context is the essential first step in understanding T-cell responses, discovering vaccine targets, and advancing T-cell-based immunotherapy programmes.
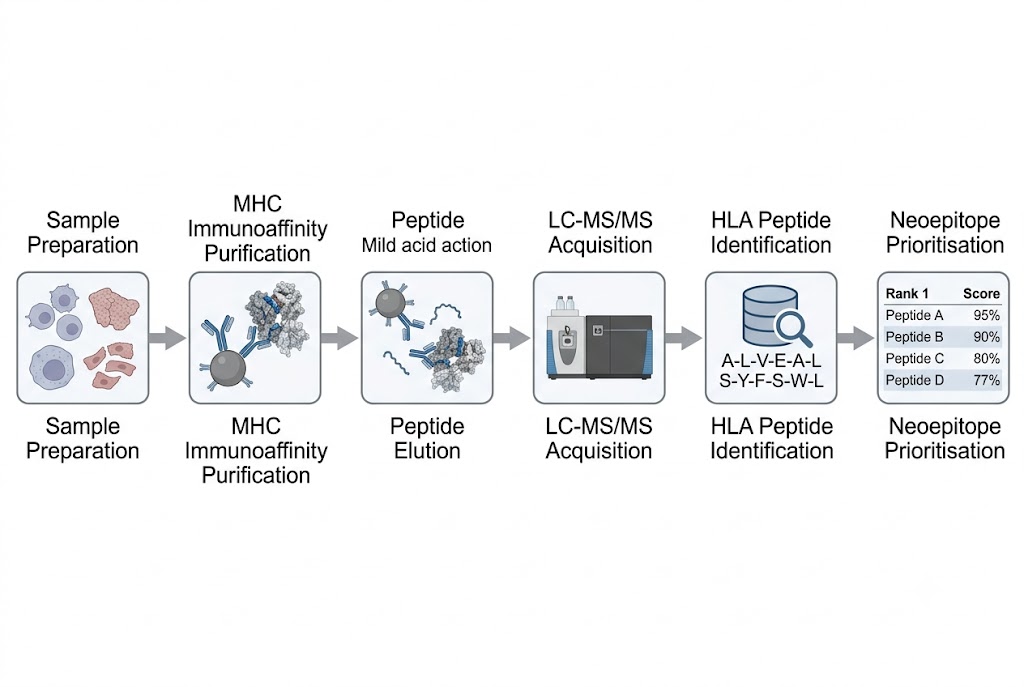
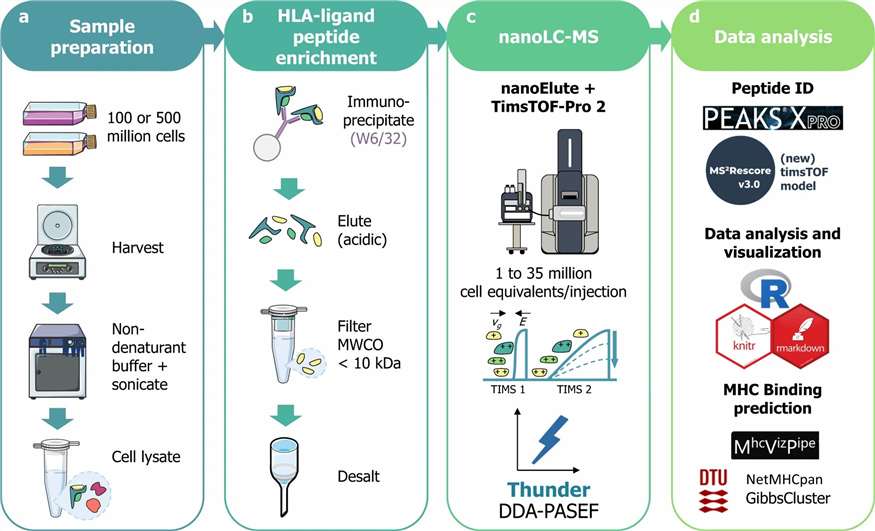
At Creative Proteomics, our Immunopeptidomics service provides end-to-end HLA ligandome profiling — from immunoaffinity-based MHC peptide enrichment through high-resolution LC-MS/MS identification to comprehensive bioinformatics analysis. We support both HLA class I (HLA-I) and HLA class II (HLA-II) workflows, processing a wide range of sample types including cell lines, primary tissues, tumour biopsies, FFPE specimens, and PBMCs.
- HLA-I and HLA-II dual capability: Dedicated workflows for class I (W6/32 antibody-based) and class II immunopeptidomics, both optimised for non-tryptic peptide identification
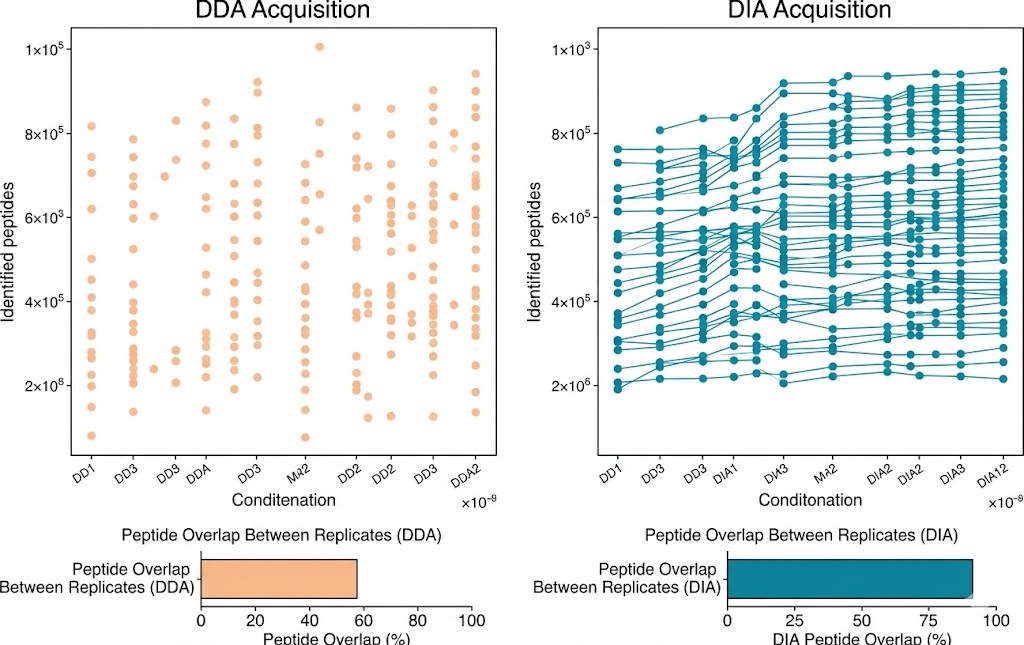
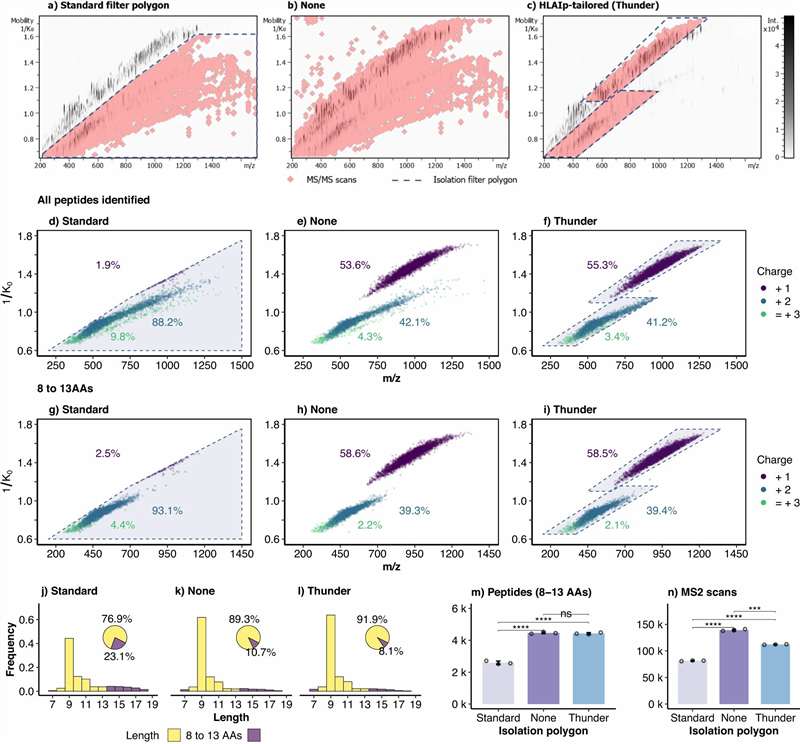
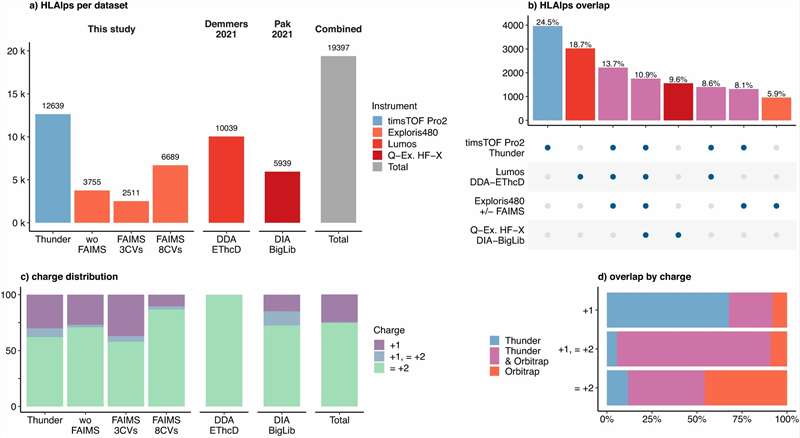
- Deep coverage with DIA/4D-DIA: Data-independent acquisition on Bruker timsTOF Pro and Orbitrap platforms extends detectable ligand depth by 30–50% over standard DDA
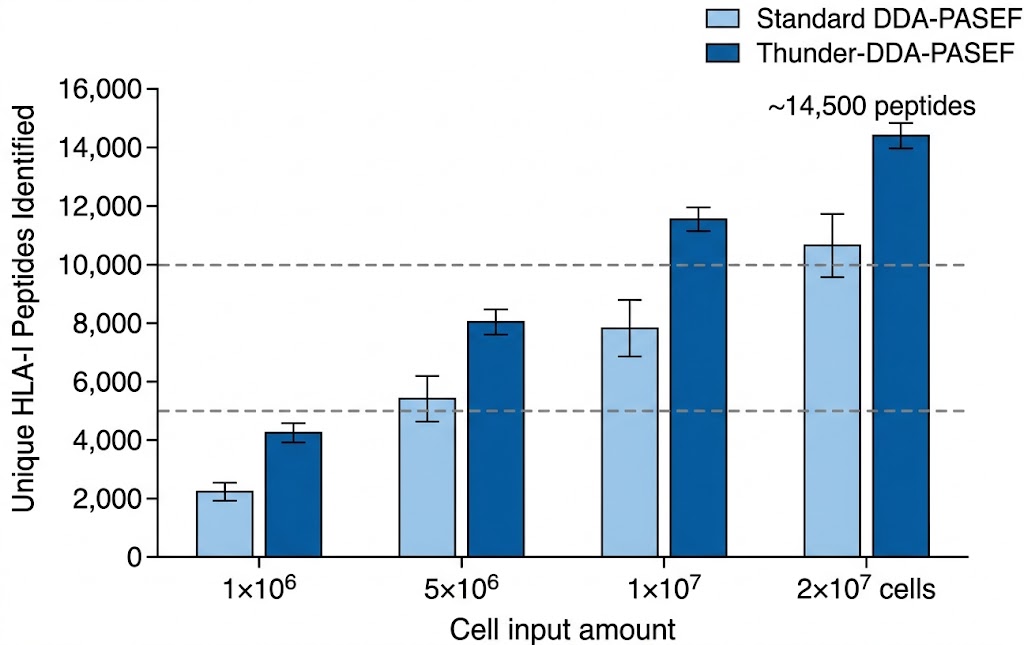
- Low-input compatibility: Validated workflows from as few as 5×106 cells; Thunder-DDA-PASEF achieves >5,000 HLA-I peptides from 1×106 cells
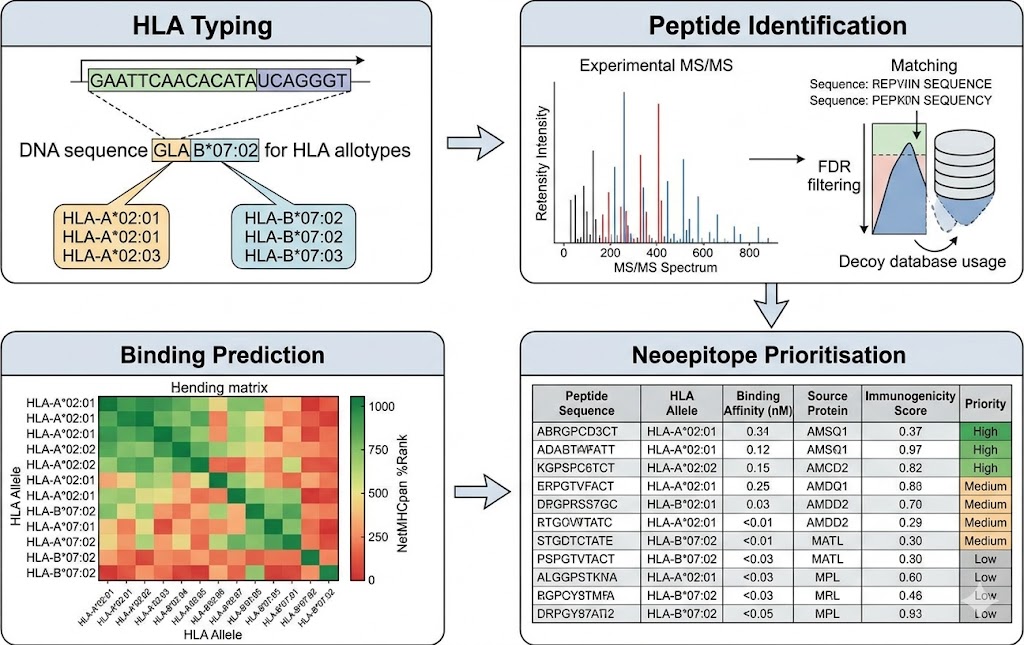
- Comprehensive bioinformatics: HLA typing, allele-specific binding prediction, multi-allelic deconvolution, and neoepitope prioritisation