CORE SERVICE
What is Proteomics-Acetyl Integrative Analysis?
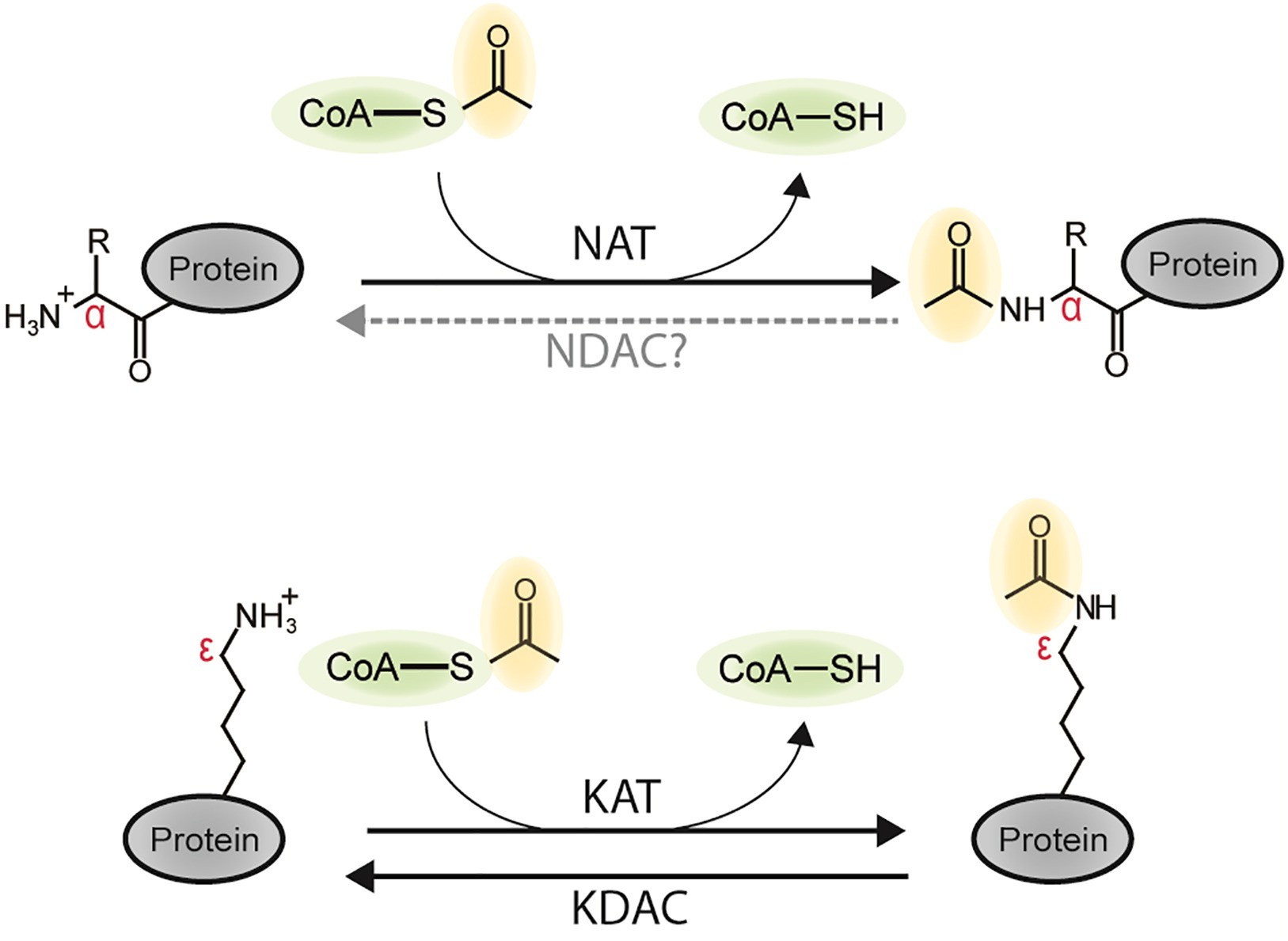
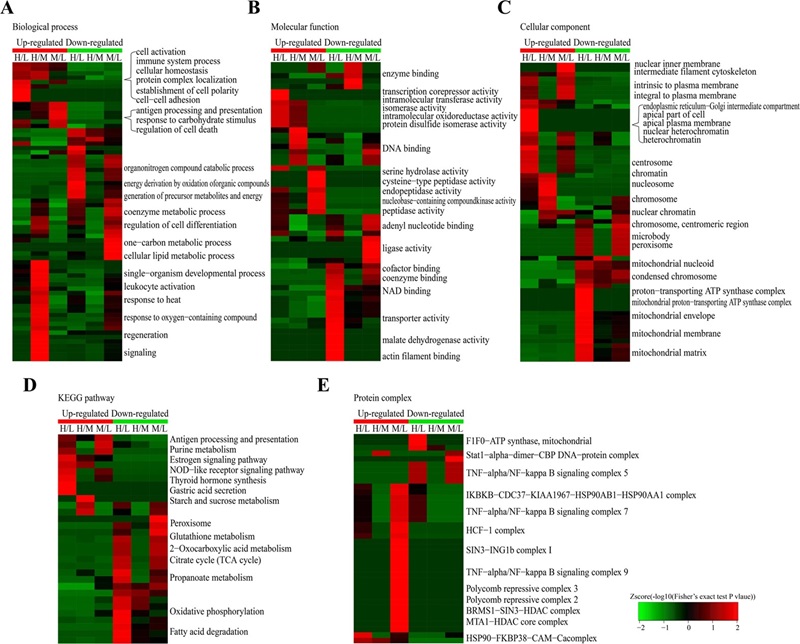
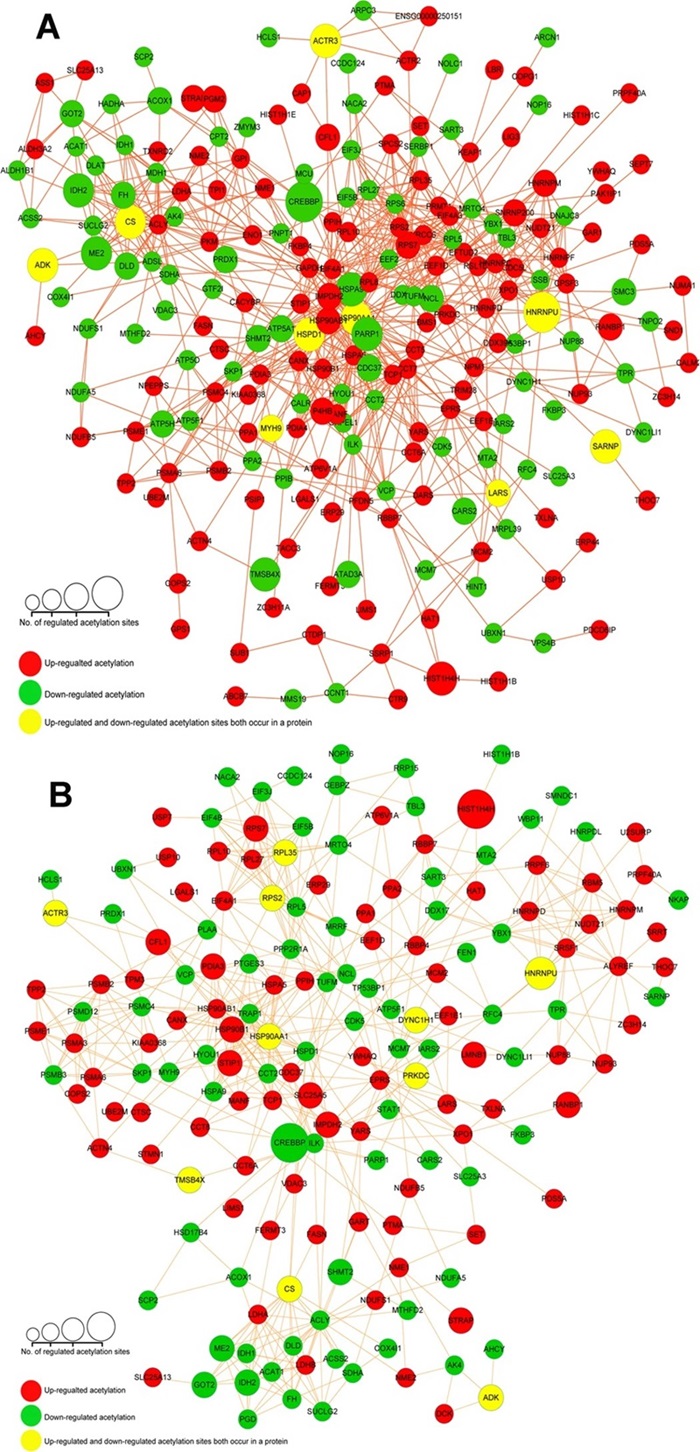
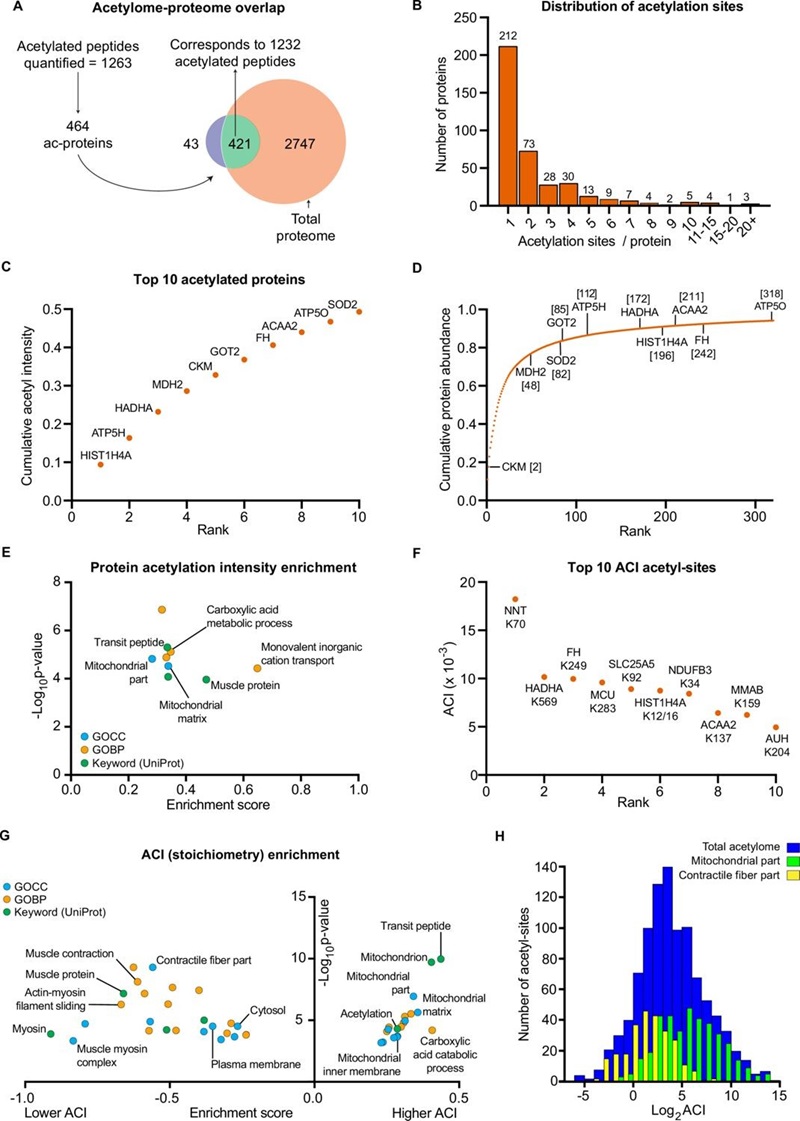
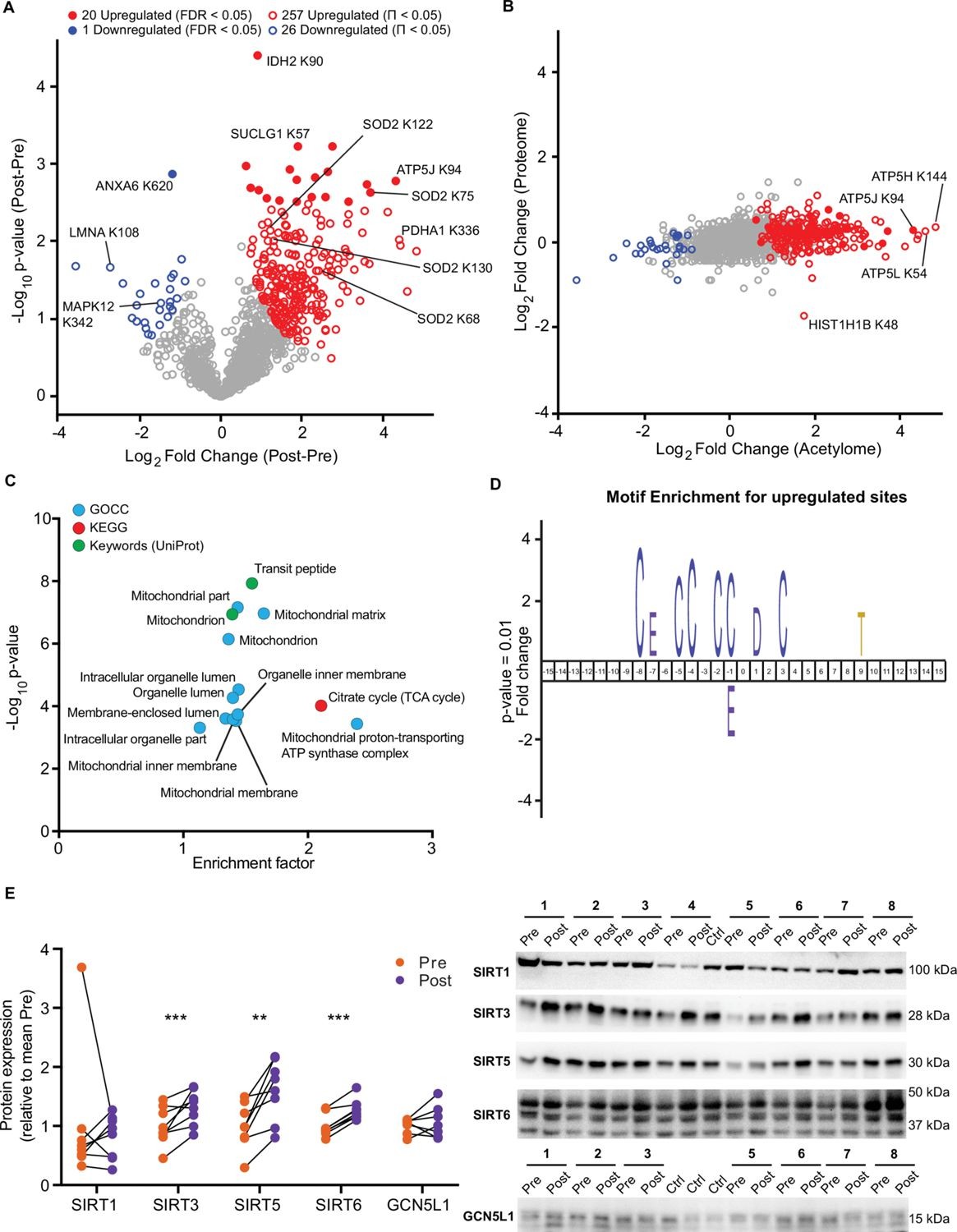
Acetylation involves the covalent addition of an acetyl group (-COCH3) to lysine residues of target proteins, primarily mediated by lysine acetyltransferases (KATs) and reversed by lysine deacetylases (KDACs). This PTM is key in epigenetic regulation, enzyme activity modulation, signal transduction, and protein-protein interaction networks.
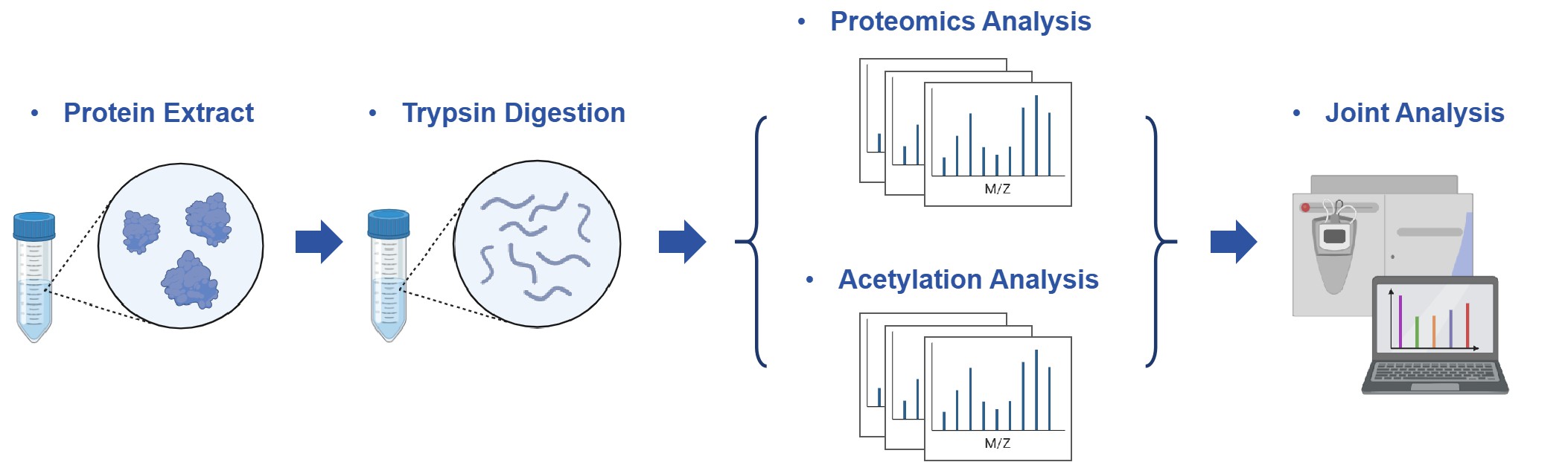
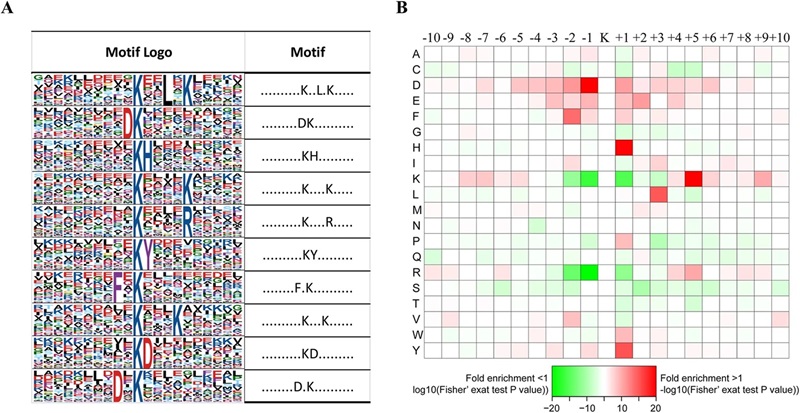
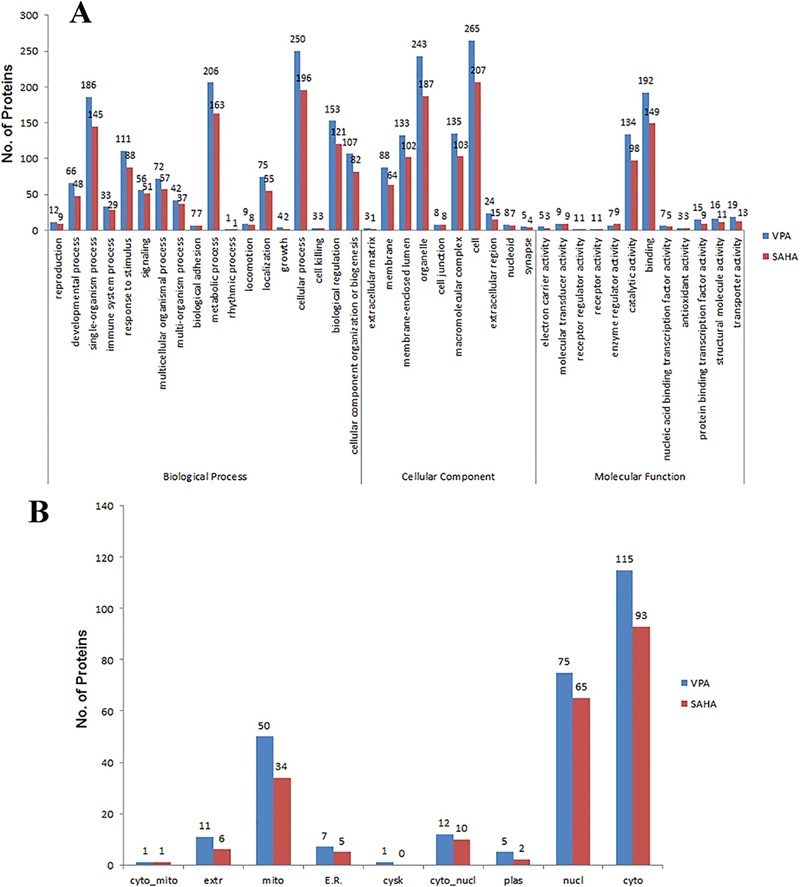
Integrative acetyl-proteomics analysis combines quantitative acetylation profiling with other omics datasets to gain deeper biological insights. By linking acetylation changes to transcript levels, metabolic fluxes, or phosphorylation dynamics, researchers can uncover regulatory mechanisms and functional pathways that are otherwise difficult to detect using single-layer analysis.