CORE SERVICE
What is the Phosphoproteome?
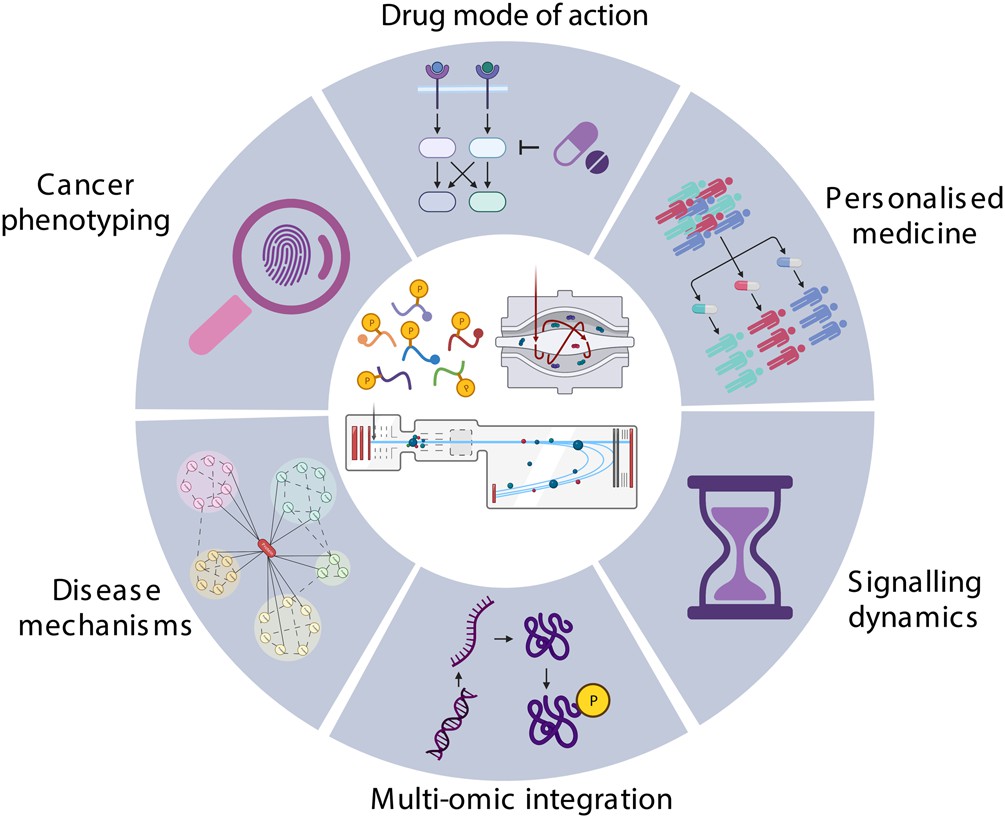
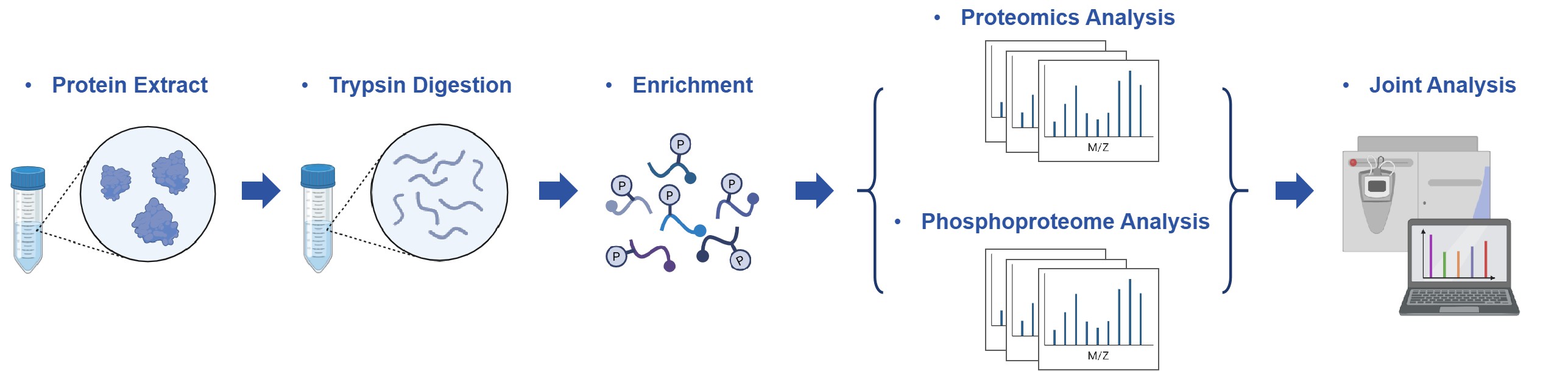
Proteins are the central executors of biological processes, and their regulation often depends on chemical modifications. Among these, phosphorylation represents one of the most dynamic and reversible events. Proteomics provides a global view of protein abundance, while phosphoproteomics reveals signaling activity by identifying phosphorylation events. Integrating these two dimensions enables researchers to understand cellular regulation's static and dynamic layers.
The phosphoproteome refers to the complete set of phosphorylated proteins in a cell, tissue, or organism at a given time. Phosphorylation is catalyzed by kinases and reversed by phosphatases. This reversible modification alters protein conformation, stability, localization, and interaction networks. Unlike total proteomics, which focuses on protein expression levels, phosphoproteomics highlights functional regulation within signaling pathways. Because phosphorylation is tightly linked to cellular responses such as proliferation, apoptosis, and immune activation, the phosphoproteome provides essential information about the dynamic state of biological systems.