CORE SERVICE
What is Precise Quantification of Histone H3?
Unveiling the Epigenetic Landscape
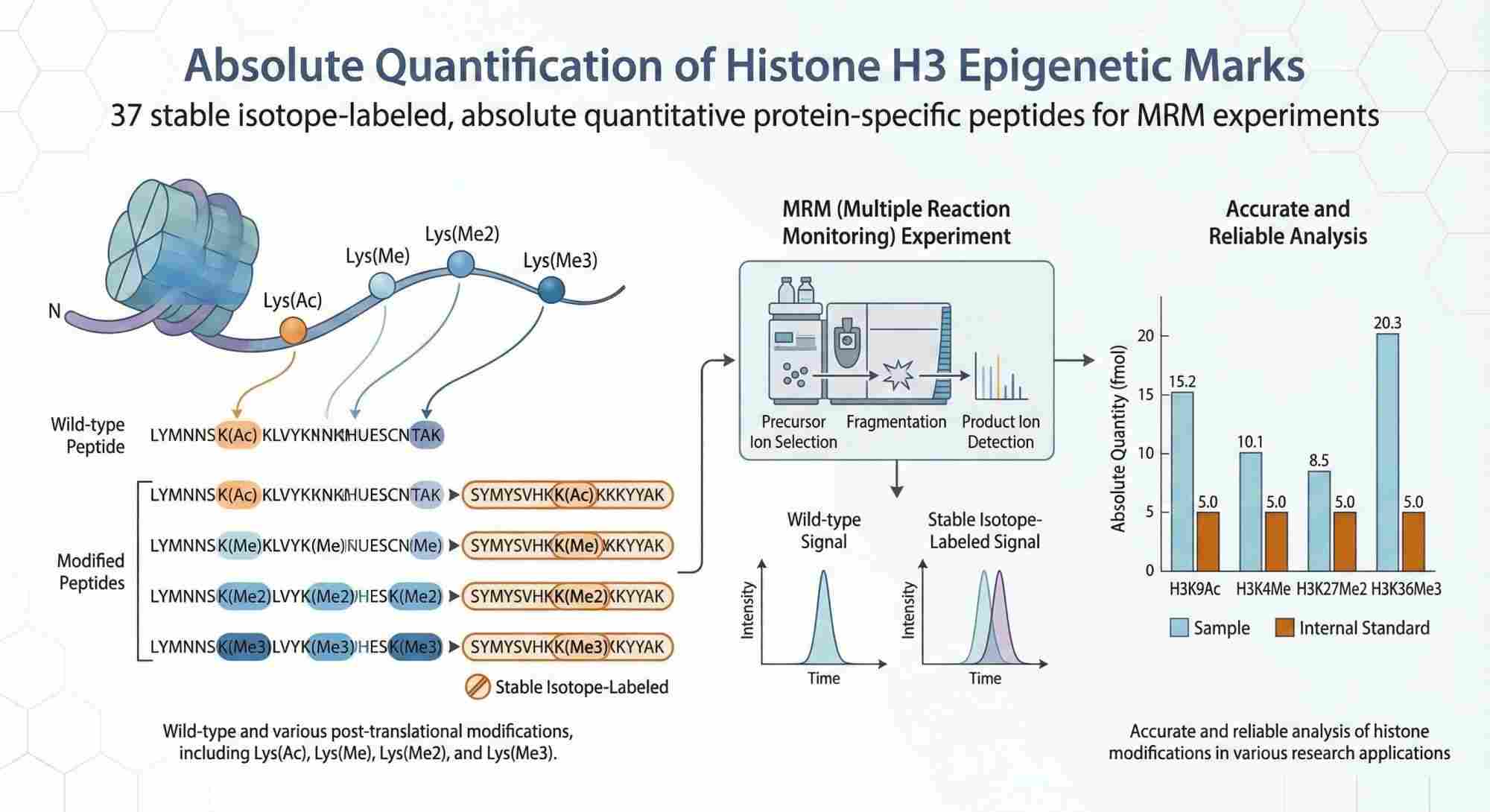
Histone H3 is a fundamental structural component of eukaryotic chromatin, forming one-half of the core tetramer within the nucleosome octamer around which DNA is wrapped. While its globular domain is essential for nucleosome stability and DNA packaging, its biological significance is predominantly defined by its unstructured N-terminal tail, which protrudes from the nucleosome core.
This tail serves as a dynamic signaling hub, subject to a vast array of covalent post-translational modifications (PTMs) such as methylation, acetylation, and phosphorylation. These modifications constitute a "Histone Code" that regulates the interaction between DNA and nuclear proteins, thereby controlling the physical accessibility of the genome.
The importance of Histone H3 extends to the regulation of nearly all DNA-templated processes, including transcription, replication, and repair. By recruiting specific "reader" proteins, H3 modifications dictate the transition between tightly packed, transcriptionally silent heterochromatin (e.g., marked by H3K9me3) and accessible, active euchromatin (e.g., marked by H3K4me3 or H3K27ac). Consequently, Histone H3 acts as a critical epigenetic determinant of cell fate, governing stem cell differentiation and developmental timing. Dysregulation of H3 PTM stoichiometry is a primary driver in oncology and metabolic diseases, highlighting its central role in maintaining genomic integrity and cellular homeostasis.
In pathological states like cancer or metabolic dysfunction, the global stoichiometry of these H3 marks shifts. However, because H3 tails often carry combinatorial modifications (e.g., phosphorylation adjacent to methylation), traditional antibodies frequently fail due to epitope occlusion or cross-reactivity. Absolute quantification via mass spectrometry is the only method capable of resolving these complex proteoforms with stoichiometric accuracy, enabling researchers to accurately decipher the "Histone Code" and its dynamic role in transcriptional regulation, DNA repair, and epigenetic inheritance.