Why Quantitative Protein Interaction Analysis Matters?
Protein-protein interactions (PPIs) are dynamic and context-dependent. Changes in interaction strength or composition often reflect pathway activation, disease states, or drug-induced perturbations. Traditional qualitative approaches frequently fail to distinguish specific interactors from nonspecific background proteins, limiting biological interpretation.
Quantitative proteomics-based PPI analysis addresses this limitation by enabling direct comparison between experimental and control conditions. In drug discovery, this is particularly crucial for target validation, mechanism-of-action studies, and profiling of off-target interactions. SILAC-based CoIP-MS has become a preferred strategy for high-confidence interaction analysis in complex biological systems.
What Is SILAC-based CoIP-MS?
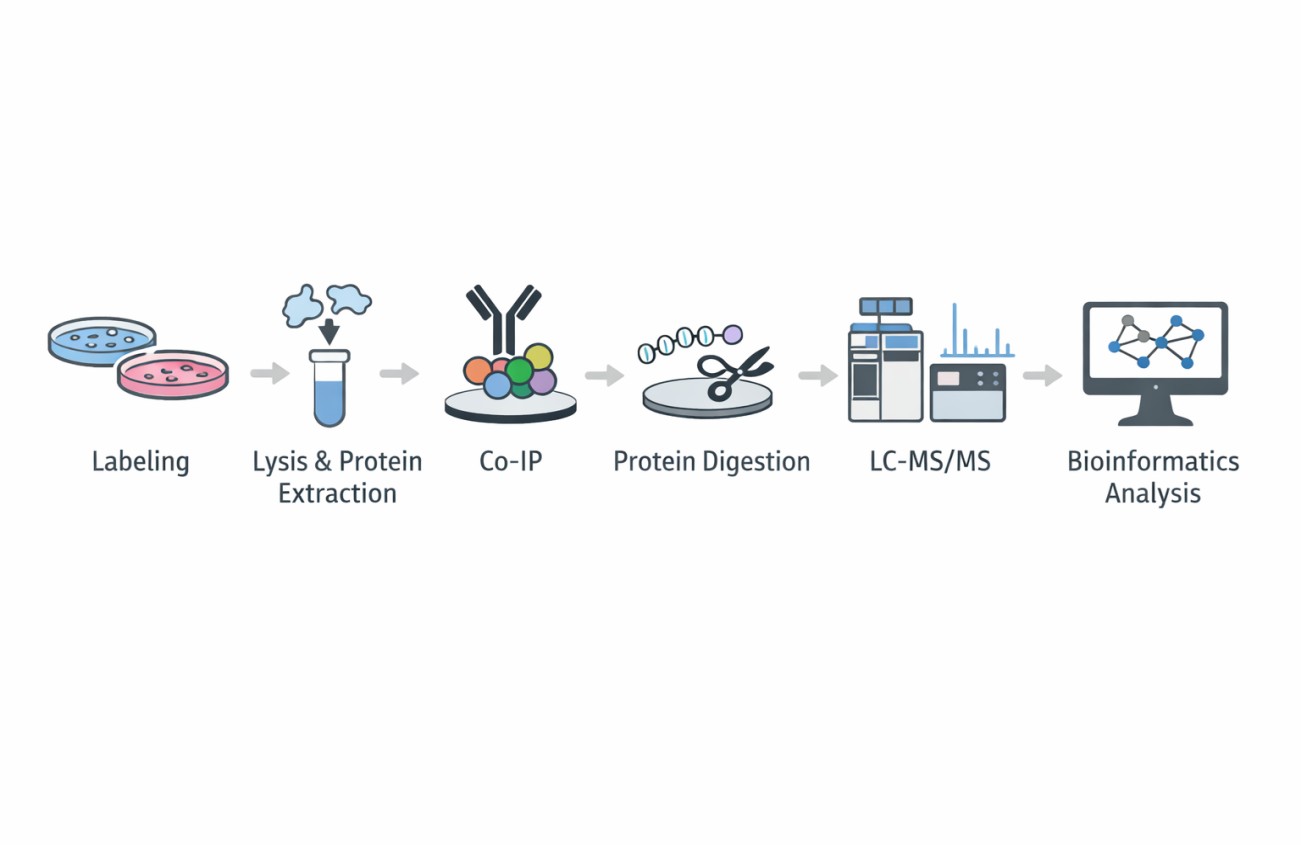
Stable Isotope Labeling by Amino acids in Cell culture (SILAC) is a metabolic labelling technique in which cells incorporate isotopically labelled amino acids during protein synthesis. When paired with co-immunoprecipitation (CoIP) and LC-MS/MS, SILAC enables accurate relative quantification of proteins co-enriched with a target bait protein.
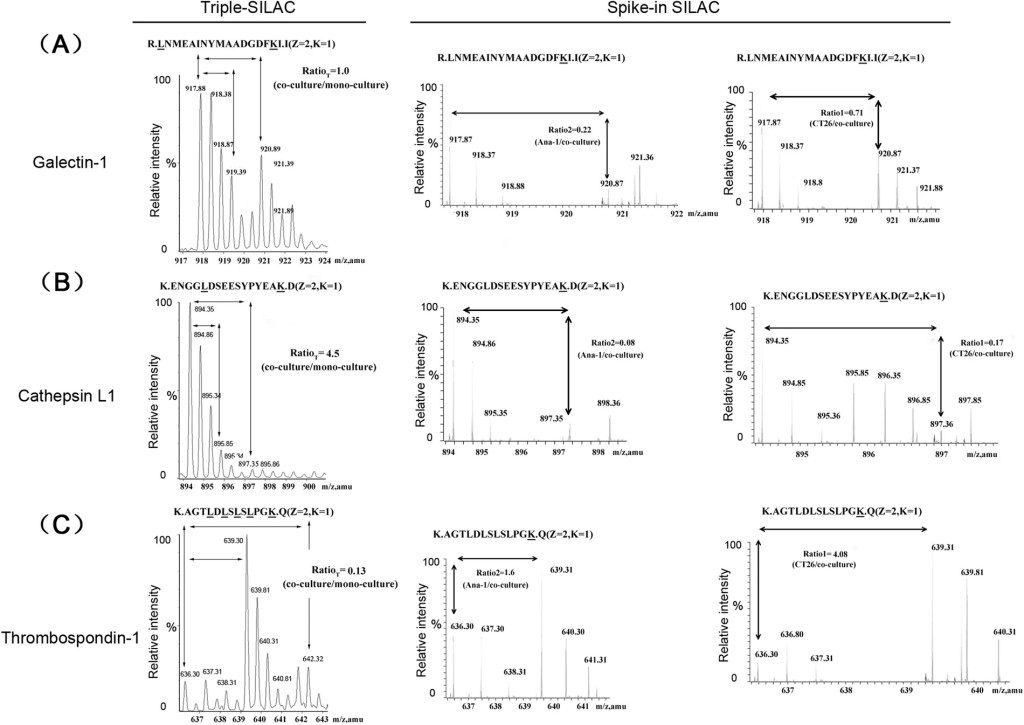
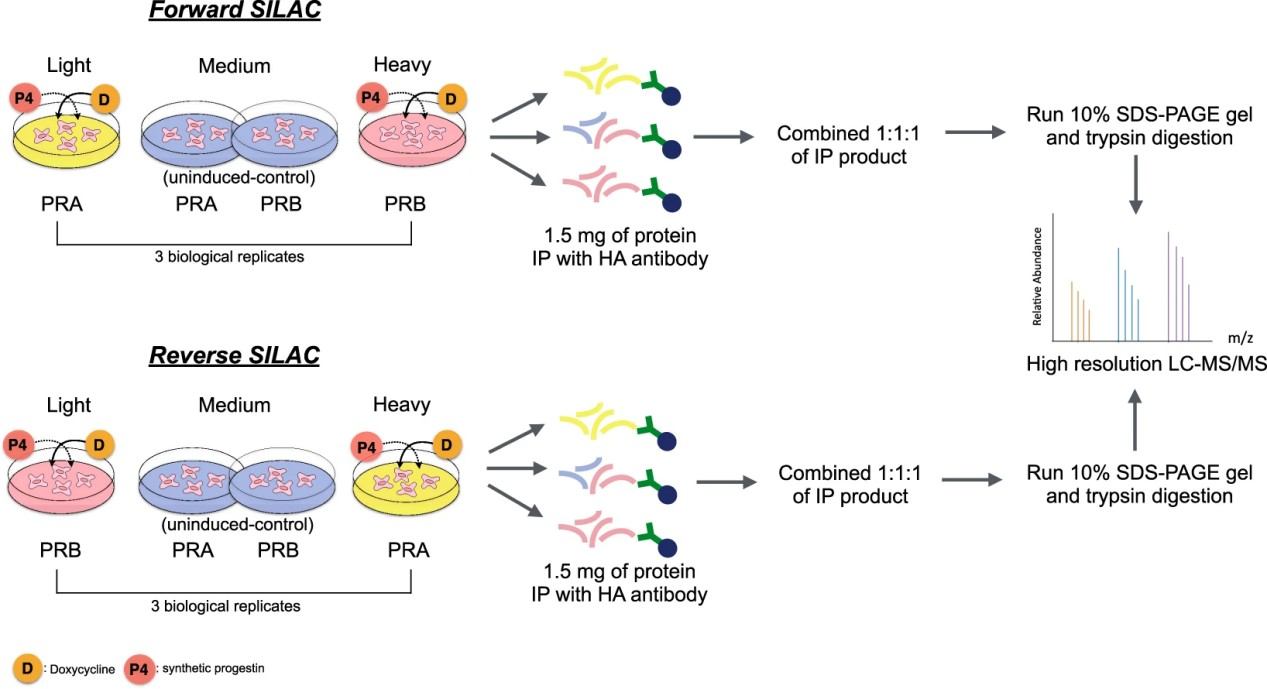
In a typical SILAC-based CoIP-MS experiment, experimental and control cell populations are differentially labelled with "heavy" and "light" amino acids. After immunoprecipitation of the protein of interest, samples are mixed early in the workflow and analysed together by mass spectrometry. The resulting heavy-to-light peptide ratios enable a direct, internal comparison of protein abundance, allowing for confident discrimination between specific interactors and nonspecific binders. This approach is widely recognised as a benchmark method for quantitative interactomics.
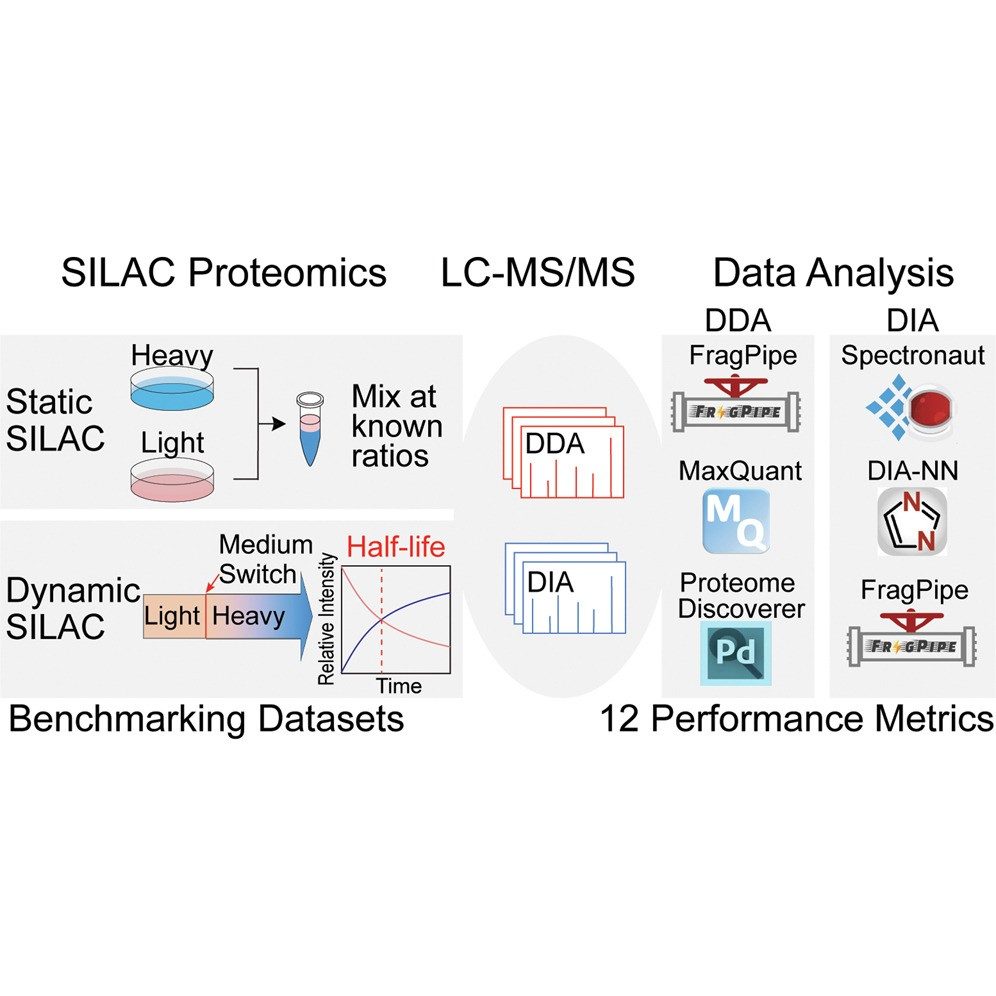
Figure 1. SILAC proteomics workflows and data analysis platforms (Frankenfield A M, et al. 2025).
Advantages of SILAC-based Proteomics
- In vivo labelling: SILAC is ideal for studies that analyse protein interactions and changes in the natural environment.
- High labelling efficiency and stability: SILAC can label almost all proteins in the cell, typically up to 99%.
- High sensitivity: SILAC can detect and quantify low-abundance proteins and is suitable for the identification of low-expressed proteins.
- Dynamic proteome analysis: SILAC is well-suited to studying the dynamics of the proteome under different conditions or time points.
Why Combine SILAC with CoIP-MS?
The integration of SILAC with CoIP-MS offers several critical methodological advantages over conventional IP-MS or post-lysis chemical labelling strategies:
- Reduced Experimental Bias: Isotope labelling occurs during cell growth, prior to protein extraction, minimising variability introduced by sample handling and processing.
- Early Sample Mixing: Heavy and light samples are combined immediately after immunoprecipitation, ensuring identical downstream processing and high quantitative precision.
- Physiological Relevance: Protein interactions are captured in living cells, preserving native conformations and post-translational modifications.
- Low False-Positive Rates: Quantitative comparison against controls enables statistical filtering of nonspecific background proteins.
Comparison with Other Quantitative Proteomics Strategies
TMT (Tandem Mass Tag) labeling enables the simultaneous comparison of multiple samples, making it ideal for large-scale studies or experiments that require high-throughput analysis. Unlike SILAC, TMT labels proteins after digestion, which can sometimes introduce variability in quantification; however, it works well with samples that cannot be metabolically labelled, such as primary tissues.
iTRAQ (Isobaric Tags for Relative and Absolute Quantitation) functions similarly to TMT, chemically tagging peptides post-digestion for simultaneous comparison across samples. iTRAQ is useful for analyzing multiple conditions or treatments in a single experiment, but can be more sensitive to technical variability in sample preparation. SILAC introduces the label during cell growth, so heavy and light samples are mixed early in the process. This reduces experimental error, preserves native protein states, and provides more reliable relative quantification, especially for detecting subtle changes in protein interactions.