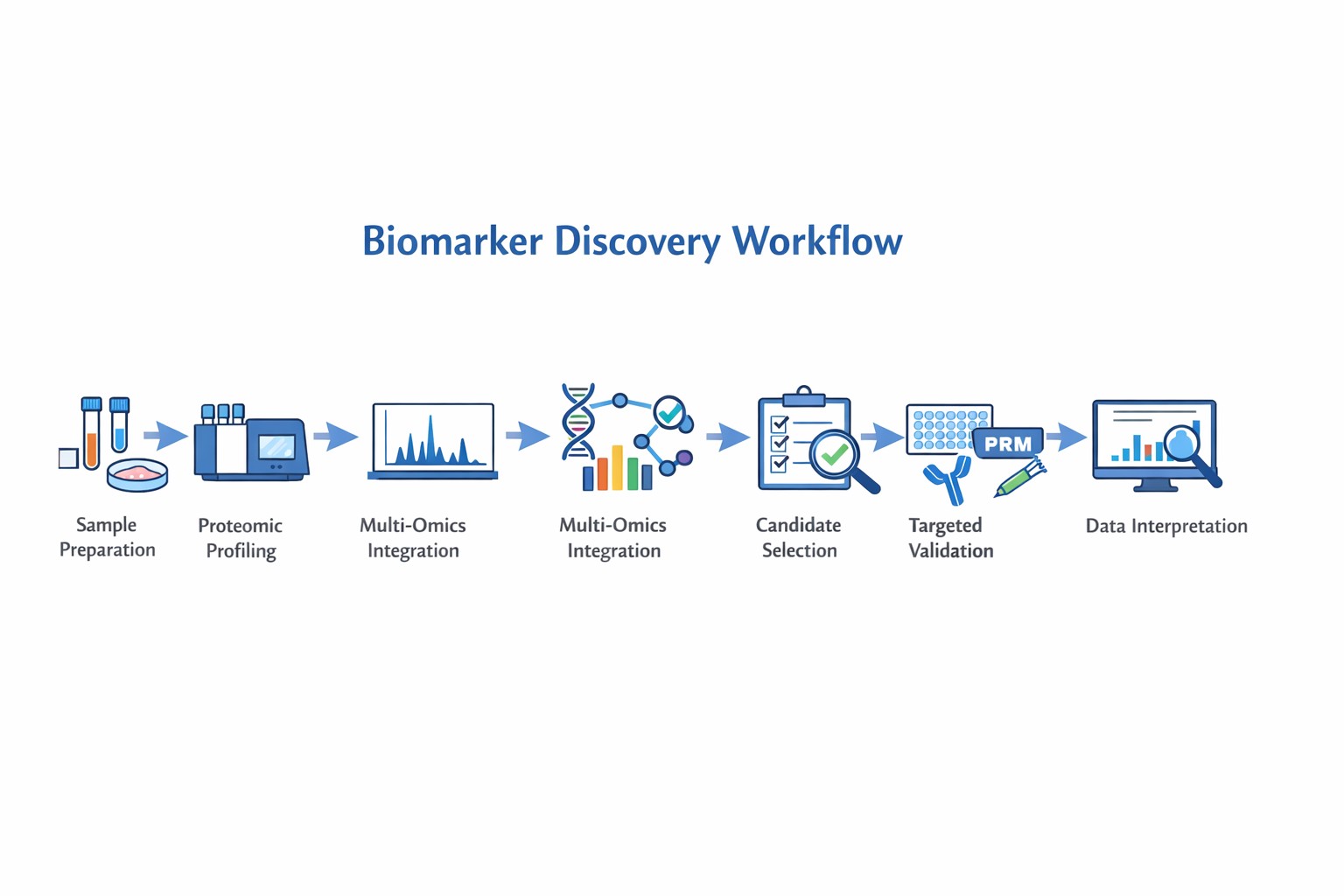
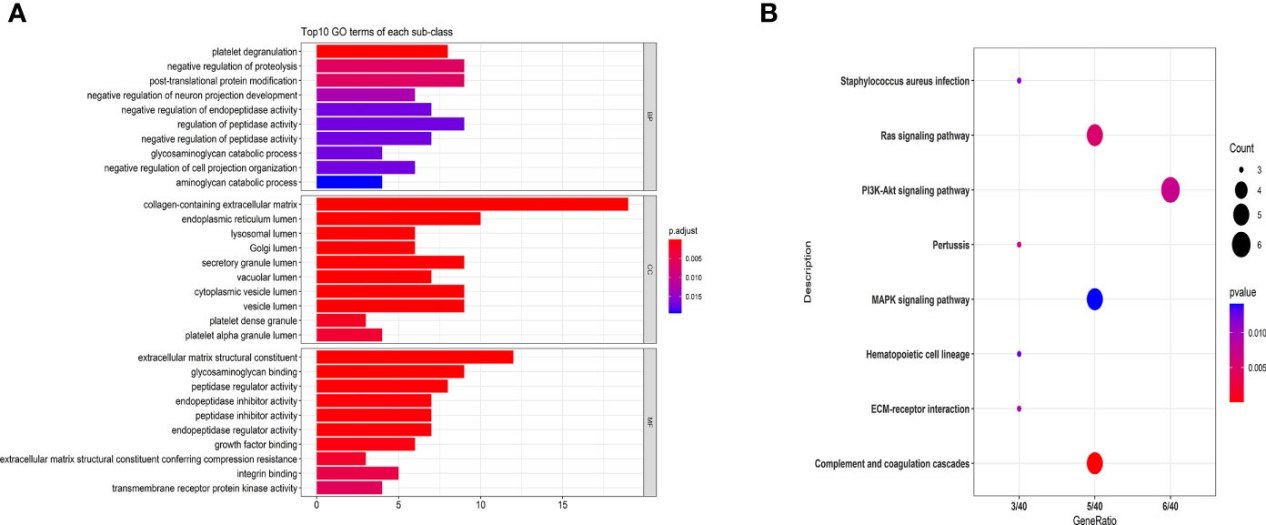
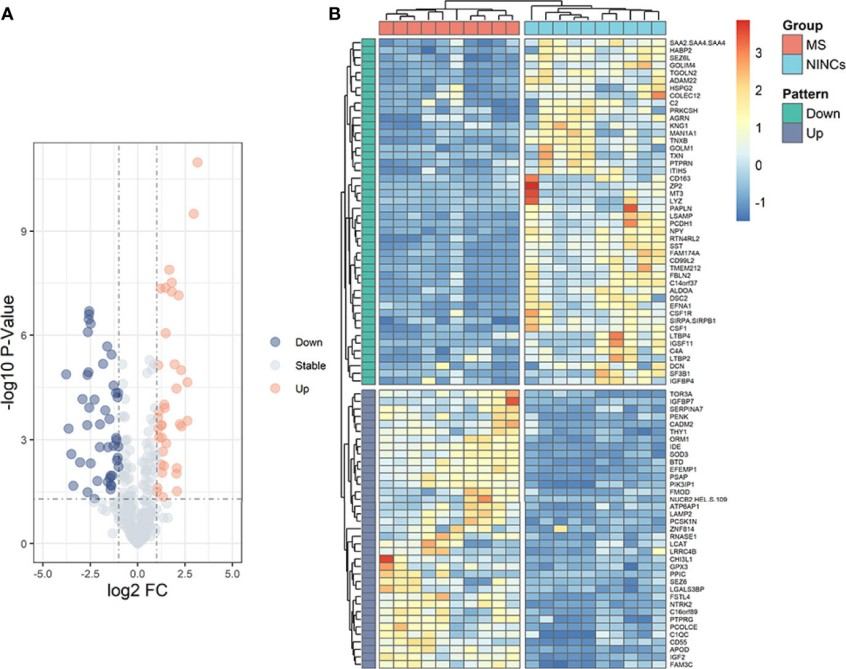
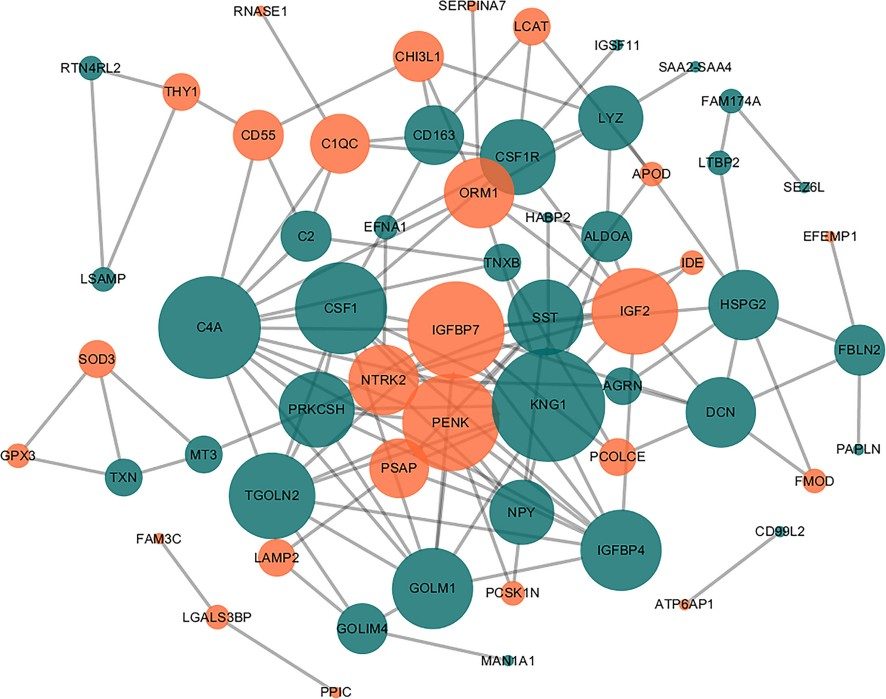
Why Biomarker Identification is Important?
Biomarkers are measurable biological indicators that reflect physiological states, pathological processes, or responses to interventions. They provide essential insights into molecular events and disease mechanisms. Accurate biomarker identification is pivotal for life science research and translational studies. Biomarkers can span nucleic acids, proteins, metabolites, epigenetic modifications, and cellular components.
Reliable biomarkers are characterized by specificity and reproducibility. They often correlate with disease progression or distinct biological phenotypes. In the context of precision research, biomarkers guide the selection of targets for molecular studies, stratify biological samples, and enhance understanding of complex molecular networks.
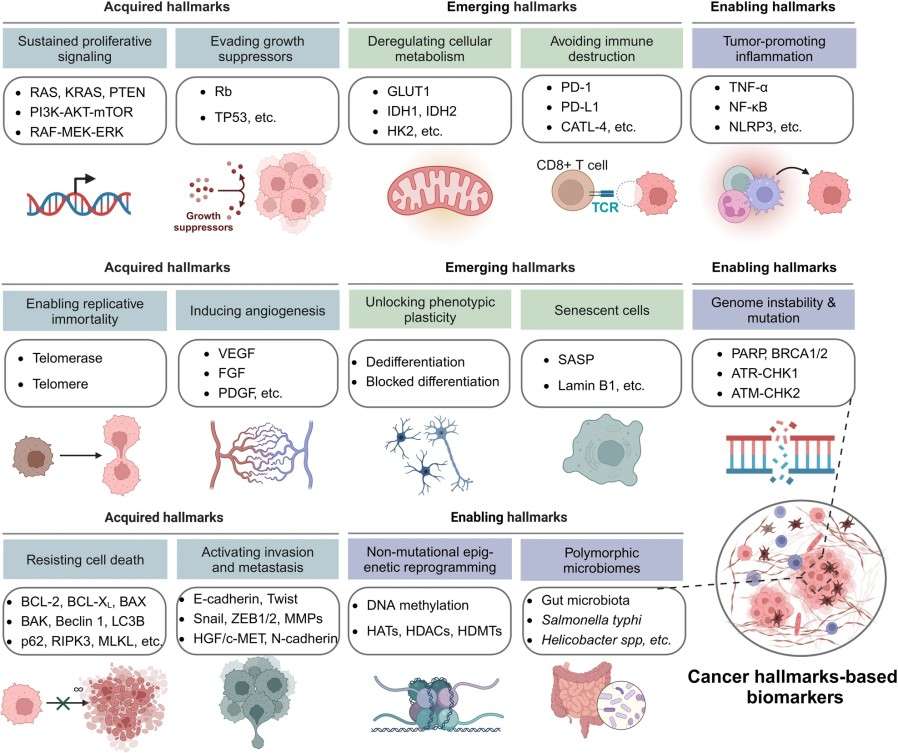
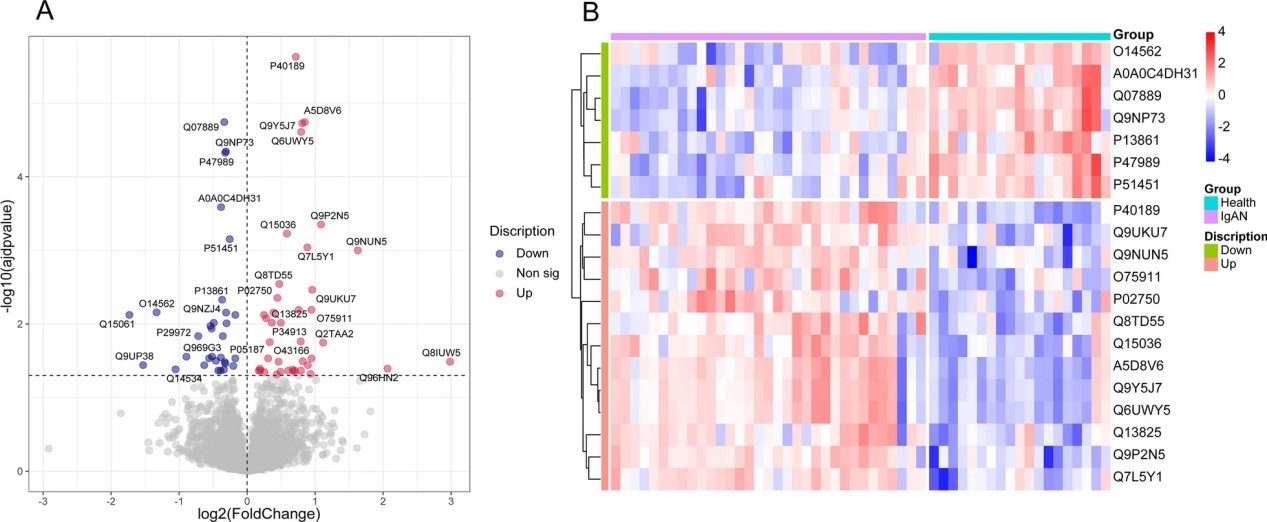
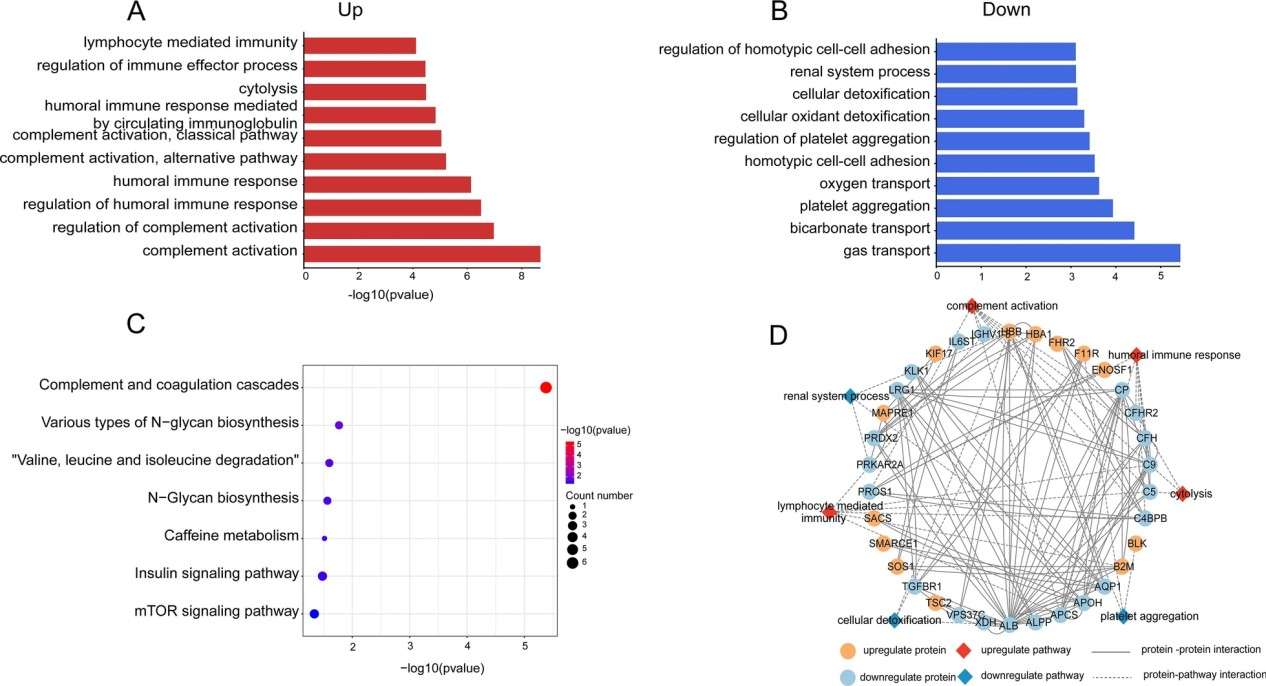
Figure 1. The 14 cancer hallmarks-based biomarkers (Zhou Y, et al., 2024).
MS-Based Proteomics for Biomarker Identification
High-Resolution Protein Profiling
Mass spectrometry enables researchers to simultaneously measure the abundance, structure, and modifications of thousands of proteins. High-resolution instruments generate precise and reproducible protein data. These tools can detect even proteins present at very low levels, which are often the most important biomarkers.
Quantitative Accuracy
Quantitative proteomics strategies enable the accurate comparison of protein levels across different conditions or patient groups. Methods like label-free quantification or stable isotope labeling provide clear numerical values for protein abundance.
Detection of PTMs
Proteins often undergo chemical changes after they are produced, such as phosphorylation or glycosylation. Mass spectrometry can detect these modifications, revealing functional changes that may indicate disease processes.
Quantitative Proteomics Strategies for Biomarker Discovery
Quantitative proteomics provides the foundation for identifying reliable biomarkers by accurately measuring protein abundance across biological samples.
- Label-free quantification measures protein abundance directly from the intensity of peptide ions detected by mass spectrometry. Spectral counting estimates protein abundance by counting the number of MS/MS spectra assigned to peptides of a given protein.
- SILAC incorporates heavy isotope-labeled amino acids into the proteome of cultured cells. After mixing labeled and unlabeled samples, MS analysis quantifies relative protein abundance based on peptide ion intensity ratios. SILAC offers high accuracy and low technical variability, making it an ideal tool for mechanistic studies in cell culture.
- TMT and iTRAQ are chemical labeling strategies applied to peptides after protein digestion. These isobaric tags allow multiplexing of multiple samples in a single MS run, increasing throughput and experimental consistency.