
N-Terminal Sequencing
Protein C-Terminal Sequencing
Full-Length Sequencing
Protein De Novo Sequencing
Antibody De Novo Sequencing
Peptide Sequencing
Proteomics, the study of proteins and their functions within biological systems, has become a pivotal field in modern molecular biology. This article explores advanced protein sequencing techniques, shedding light on N-Terminal Sequencing, Full-Length Sequencing, Edman Degradation, Protein De Novo Sequencing, C-Terminal Sequencing, Top-Down Protein Sequencing, Antibody De Novo Sequencing, and Peptide Sequencing.
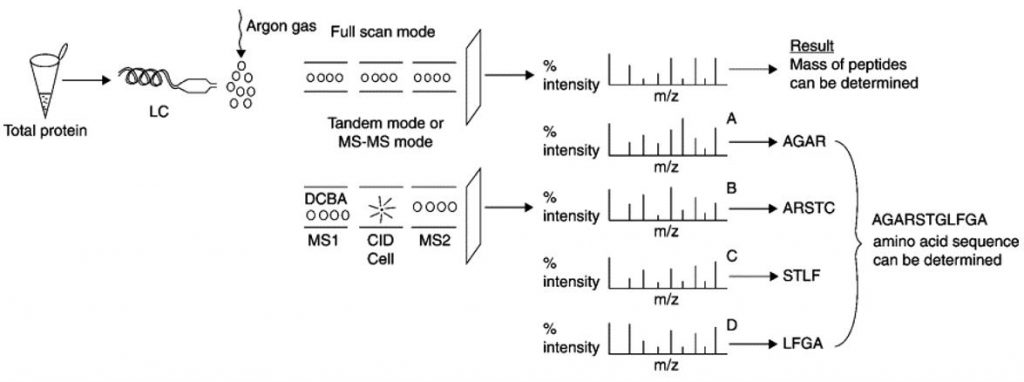
Protein sequencing using tandem MS(Saraswathy et al., 2011).
N-Terminal Sequencing
The N-terminus refers to the end of a polypeptide chain where the first amino acid is located. N-terminal sequencing aims to identify this initial amino acid and, in some cases, adjacent amino acids. Understanding the N-terminus is essential for elucidating the primary structure of a protein and provides insights into its function within a biological context.
Edman degradation N-terminal sequencing:
One of the classic methods for N-terminal sequencing is Edman Degradation. This chemical process involves selectively removing the N-terminal amino acid, which is then subjected to identification and quantification. This cyclic process can be repeated to sequence successive amino acids. While Edman Degradation has been widely used, it has limitations, such as the size of the protein that can be analyzed and the potential for side reactions.
Mass spectrometry N-terminal sequencing:
Modern N-terminal sequencing often utilizes mass spectrometry, a powerful technique that has revolutionized proteomics. In this approach, proteins are enzymatically digested into peptides, and the N-terminal peptides are then analyzed. Mass spectrometry allows for high-throughput sequencing, providing rapid and accurate identification of N-terminal amino acids. Additionally, advancements in tandem mass spectrometry have improved the sensitivity and resolution of N-terminal sequencing.
Applications and Significance of N-Terminal Sequencing
- Protein Structure and Function: Understanding the N-terminus is crucial for predicting the folding and structural features of proteins, influencing their overall function.
- Post-Translational Modifications: Identification of N-terminal amino acids is essential for studying post-translational modifications that may occur at the protein's N-terminus, impacting its activity and localization.
- Drug Development: Knowledge of the N-terminus is valuable in drug development, particularly in designing targeted therapeutics that interact with specific regions of a protein.
Protein C-Terminal Sequencing
Protein C-terminal sequencing is a specialized analytical technique employed in the field of proteomics to determine the specific amino acid sequence at the carboxyl (C) terminus of a protein. The C-terminus plays a crucial role in protein function, post-translational modifications, and interactions with other biomolecules. Accurate identification of this terminal sequence is essential for understanding the functional properties of a protein and its involvement in various cellular processes.
The process typically involves the enzymatic or chemical cleavage of the protein at its C-terminus, generating smaller peptide fragments. These fragments are then subjected to analytical methods such as mass spectrometry, Edman degradation, or other sequencing techniques. Mass spectrometry is particularly prevalent in modern C-terminal sequencing due to its sensitivity and ability to provide information about the mass of individual amino acids.
In recent years, advancements in mass spectrometry instrumentation and associated bioinformatics tools have significantly improved the efficiency and reliability of C-terminal sequencing. The technique has found widespread application in the characterization of post-translational modifications, identification of proteolytic cleavage sites, and elucidation of protein structures.
Protein C-terminal sequencing is instrumental in understanding the biological functions of proteins and unraveling complex cellular pathways. It is especially valuable when studying proteins involved in signaling cascades, as modifications or interactions at the C-terminus often regulate protein activity.
Full-Length Sequencing
Protein full-length sequencing provides a holistic view of a protein's primary structure, enabling researchers to decipher the entire sequence of amino acids. This information is vital for understanding how proteins fold, function, and interact with other molecules.
Knowing the full sequence allows researchers to predict and study the secondary and tertiary structures of proteins. This is particularly important in structural biology and drug design, where detailed knowledge of a protein's architecture is essential.
Mass Spectrometry-Based Full-Length Sequencing Methods:
- Bottom-Up Proteomics: In this approach, proteins are enzymatically digested into peptides, and the resulting peptides are analyzed using mass spectrometry. Software tools then piece together the peptide sequences to reconstruct the full-length protein sequence.
- Top-Down Proteomics: Top-down sequencing involves analyzing intact proteins directly, providing information about the full sequence and potential post-translational modifications. High-resolution mass spectrometers are crucial for the success of top-down proteomics.
Protein De Novo Sequencing
Protein de novo sequencing is a cutting-edge analytical technique in the field of proteomics that involves determining the amino acid sequence of a protein without prior knowledge of its genetic code or reference database. Unlike traditional methods that rely on database matching, de novo sequencing provides a valuable tool for characterizing novel or modified proteins.
The process begins with the acquisition of mass spectrometry data from fragmented protein ions. These spectra, often in the form of tandem mass spectrometry (MS/MS) data, contain information about the individual amino acids within the protein. Computational algorithms are then employed to interpret these spectra and infer the sequence of amino acids, allowing researchers to reconstruct the original protein sequence.
The challenges associated with de novo sequencing arise from the complexity of mass spectrometry data and the potential for ambiguous interpretation. Researchers utilize advanced algorithms and bioinformatics tools to address these challenges, iteratively refining the predicted sequence through comparisons with experimental data. Additionally, advancements in mass spectrometry instrumentation and computational techniques have enhanced the accuracy and reliability of de novo sequencing.
Protein de novo sequencing has proven particularly valuable in the study of post-translational modifications, rare protein variants, and in situations where reference databases are limited or nonexistent. By enabling the elucidation of protein sequences independent of genomic information, de novo sequencing contributes significantly to our understanding of the proteome and opens avenues for the discovery of novel biomarkers and therapeutic targets.
Antibody De Novo Sequencing
Antibody de novo sequencing is an advanced analytical technique within the realm of immunoproteomics that focuses on determining the precise amino acid sequence of an antibody without relying on existing genetic information or reference databases. Antibodies, also known as immunoglobulins, play a pivotal role in the immune system by recognizing and binding to specific antigens.
The process begins with the isolation and purification of the target antibody, followed by enzymatic or chemical cleavage to generate smaller peptide fragments. Mass spectrometry, often coupled with high-performance liquid chromatography (HPLC), is then employed to analyze these fragments and deduce the amino acid sequence. The unique challenge in antibody de novo sequencing arises from the complexity of immunoglobulin structures, including variable regions that exhibit significant diversity.
Computational algorithms and bioinformatics tools are integral to interpreting mass spectrometry data and reconstructing the antibody sequence accurately. This method is especially valuable for the characterization of monoclonal antibodies, therapeutic antibodies, or antibodies with post-translational modifications. By elucidating the antibody's primary structure, researchers gain insights into its binding specificity, affinity, and potential modifications, ultimately facilitating the design and development of more effective antibody-based therapies and diagnostics.
Peptide Sequencing
Peptide sequencing refers to the determination of the linear order of amino acids in a peptide chain. This process is essential for understanding the primary structure of proteins. Peptide sequencing is a key step in proteomic analysis, providing a snapshot of the protein composition. It serves as a bridge between the identification of peptides and the elucidation of complete protein structures.
Techniques and Tools of Peptide Sequencing
Mass Spectrometry: Mass spectrometry is a predominant technique for peptide sequencing. Peptides are ionized, and their mass-to-charge ratios are measured. Tandem mass spectrometry (MS/MS) provides fragmentation data, allowing for the determination of peptide sequences.
Edman Degradation: Edman Degradation, although traditionally used for N-terminal sequencing, can be adapted for peptide sequencing. It involves cyclic degradation of the peptide, revealing one amino acid at a time.
De Novo Sequencing: De novo sequencing is employed when the genomic or protein sequence information is unavailable. It involves the interpretation of mass spectrometric data to deduce the peptide sequence without relying on a reference database.
Reference
Saraswathy, Nachimuthu, and Ponnusamy Ramalingam. Concepts and techniques in genomics and proteomics. Elsevier, 2011.