The importance of protein modifications
Protein is the basic functional unit that performs cellular functions, and its expression is regulated by genomics and epigenetics. Proteins usually need to be modified to varying degrees to achieve the desired function after expression. The post-translational modification process is tightly regulated by a series of modified enzymes and de-modification enzymes, which allows the protein to exhibit a certain stable or dynamic specific function at a certain instant. Protein post-translational modification (PTM) increases the functional diversity of the proteome by covalently adding functional groups or proteins, regulating proteolytic cleavage of the subunit or degradation of the entire protein.
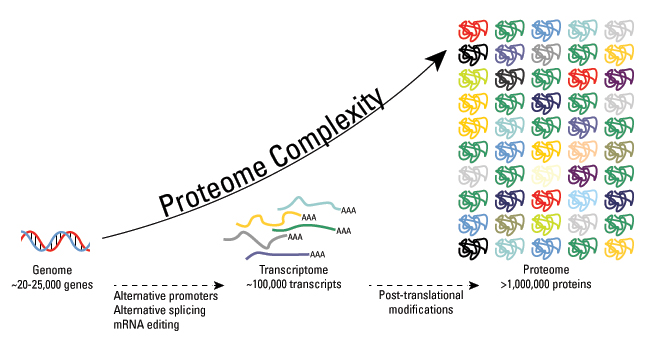
These modifications include phosphorylation, glycosylation, nitrosylation, methylation, acetylation, lipidation, and proteolysis, and affect almost all aspects of normal cell biology and pathogenesis. Protein phosphorylation is by far the most common PTM and has been detected in approximately 17,500 human gene products. Post-translational modification is a key mechanism for increasing the diversity of proteome. Although the genome contains only 20,000 to 25,000 genes, the proteome is estimated to contain more than 1 million proteins. Changes in transcription and mRNA levels increase the size of the transcriptome relative to the genome, and numerous different post-translational modification increases the complexity of the proteome relative to the transcriptome and genome. Therefore, identifying and understanding PTMs is critical to the study of cell biology and disease treatment and prevention.
Progress in Protein Modification Research
Due to the heterogeneity and relative abundance of post-translationally modified proteins, research of protein post-translational modification is mainly based on existing proteomics technology systems, including electrophoresis, chromatography, mass spectrometry, and bioinformatics tools. Or the peptide segment is enriched and separated, the heterogeneity caused by the modification is eliminated, and the modification site is marked to make a difference from the theoretical mass. The difference is detected by mass spectrometry to identify the protein, and the modification site is identified by tandem mass spectrometry. PTMs are widely present in eukaryotic cell biology and are critical for the signaling and life activities of organisms, but PTMs identification is often more difficult than identification of unmodified peptides.
Protein phosphorylation is the most common and most studied modification in organisms, and tyrosine phosphorylation, especially phosphorylation of tyrosine kinase receptors, has been shown to play a key role in the induction and growth of cancer cells. A variety of small molecule inhibitors and monoclonal antibodies against different tyrosine kinase receptors have also been developed as first-line drugs for the treatment of cancer. Currently, for well-studied post-translational modifications of proteins mainly include phosphorylation, acetylation, methylation and ubiquitination.
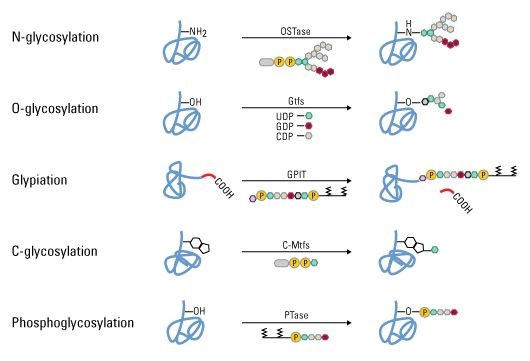
Figure 2. Some types of protein PTMs
The research methods and key technologies of phosphorylation include immunoprecipitation, flow cytometry, two-dimensional gel electrophoresis, and solid phase metal affinity chromatography. The main research methods for acetylation include identification of acetylation sites by mass spectrometry, identification of acetylation sites based on acetylated antibodies that specifically recognize acetylated lysine residues, identification of acetylation by label-based methods, modification the site and so on. The main research approaches for methylation include methylation-specific PCR, bisulfite sequencing, and high-resolution melting curve.
The research methods and key technologies of glycosylation include radiolabeling, molecular fluorescent labeling, electrophoresis, lectin labeling, antibody labeling, chemical enzyme methods and so on. The types of techniques currently applied for ubiquitin proteins are relatively monotonous. The detection of ubiquitin proteins, the localization of ubiquitin targets, and the exploration of the properties of ubiquitin proteins, these techniques must be continuously improved and modified. For example, traditional protein post-translational modification studies primarily rely on specific antibody-based immunoassays or radiolabeling techniques. These methods play an irreplaceable role in the study of cellular signaling processes mediated by post-translational modifications at a single site. However, due to some shortcomings of the above-mentioned techniques, such as high operational requirements and long preparation period of specific antibodies, it is difficult to achieve large-scale detection of post-translational modification of proteins.
Progress in Protein Modification Detection Technology
In recent years, the proteomics strategy based on liquid chromatography-mass spectrometry (LC-MS) has developed rapidly, which provides a powerful research tool for protein post-translational modification research at the system level. And mass spectrometry-based proteomics greatly gives a deeper understanding of the occurrence and dynamics of protein post-translational modification. To date, quantitative proteomics has been used primarily to study PTM regulation in cell culture models, which provides new insights into the role of abnormal PTM patterns in human disease. Mass spectrometry libraries have been widely used to accurately study the quantitative analysis of various proteins, especially in the application research of quantifying known proteins.
The new strategy of immunoaffinity preconcentration of PTM peptides with monoclonal antibodies combined with liquid chromatography-mass spectrometry (LC-MS/MS) analysis has attracted much attention in many post-translational modification studies. The basic method of PTMScan technology involves the use of 9M urea lysate to lyse diseased cell lines or patient tissue blocks (enough to extract 10 mg of protein) to obtain protein lysates, which are obtained by digestion with endonuclease (usually using trypsin). Peptide mixture, the peptide mixture was purified by C18 purification column, and then the peptides containing different protein post-modification were affinity-enriched and purified by CST company's unique protein post-translational modification motif. - Tandem mass spectrometry (LC-MS/MS) for semi-quantitative analysis.
In addition, bioinformatics analysis methods have also made progress in the field of PTM research. In recent years, with the development of proteomics technology, bioinformatics research based on proteomics, especially modified proteomics data, can provide a holistic and post-translational modification and deeper understanding. In 1999, Nikolaj Blom of the Center for Biological Sequence Analysis (CBS) used phosphorylation sites of 210 tyrosine, 584 serine and 108 threonine known at the time as a training set, and the prediction of non-specific protein phosphorylation sites was achieved for the first time by using the neural network algorithm. Since then, similar experiments have been gradually carried out, and successive studies have gradually come out. A stud which compared normal skin and skin cancer tissues in mice and combined with quantitative proteome and quantitative phosphorylation data, showed that 47.3% of proteins were found to be regulated at both protein and phosphorylation levels, while more than half of the proteins were only regulated at the regulation level. It is foreseeable that the combination of calculation and experiments, through experimental methods to verify or at least partially verify the prediction results has become a research trend in the field.