
Stable isotope labeling using amino acids in cell culture (SILAC) is a powerful method based on mass spectrometry that identifies and quantifies relative differential changes in protein abundance. First used in quantitative proteomics in 2002, it provides accurate relative quantification without any chemical derivatization or manipulation.
The Principle of SILAC
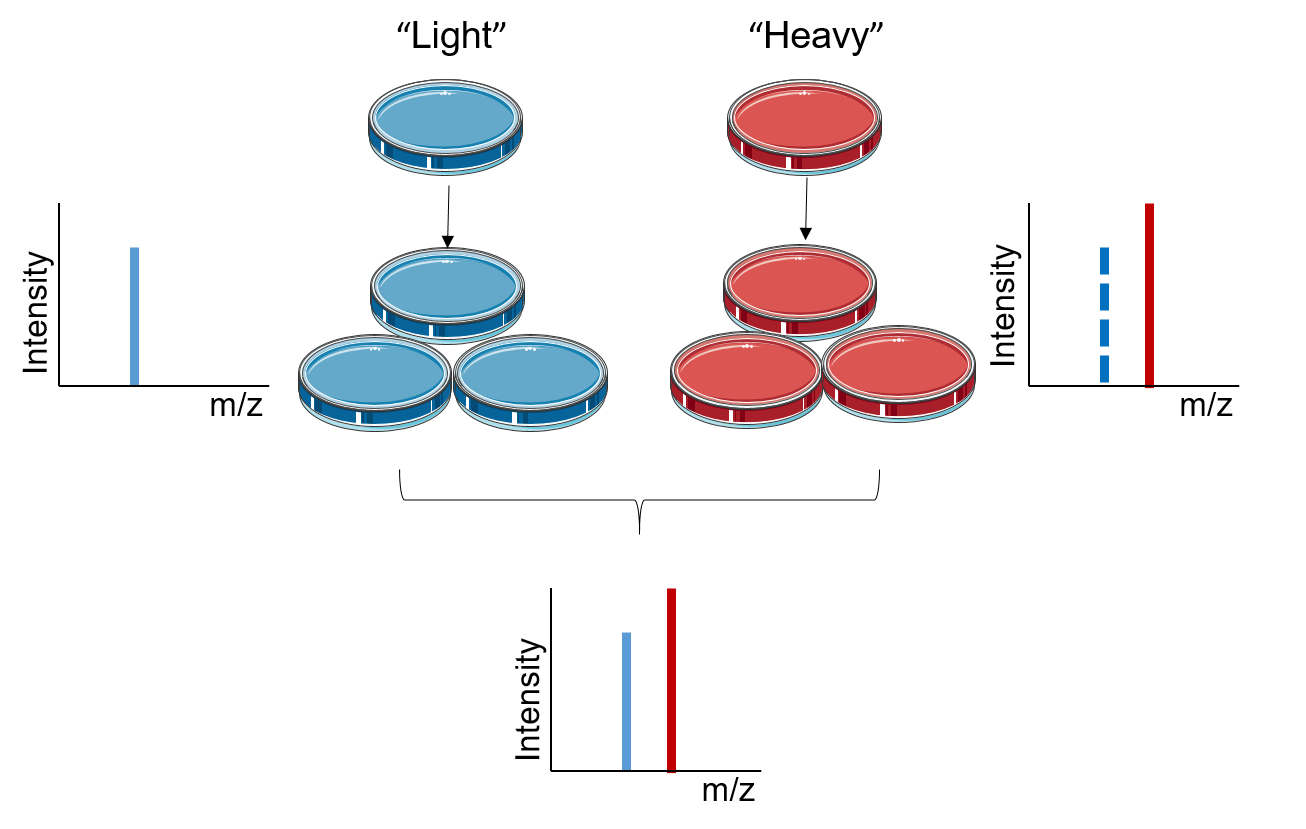
Figure 1. The principle of SILAC
The principle of SILAC is based on metabolically incorporating stable isotope labeled amino acids into the entire proteome. In SILAC, two populations of cells are grown in two different culture media, the “light” medium that contains amino acids with the natural isotope and the “heavy” medium that contains stable isotope labeled amino acids. After a sufficient number of cell divisions, all the proteins from the cells cultured in "heavy" medium contain amino acids in the heavy state. Analyzed with LC-MS/MS, the quantification of SILAC is based on testing the ratio of introduced isotope-labeled peptides to unlabeled peptides. The signal intensities from light and heavy samples allow for quantitative comparison of their relative abundances in the mixture.
The Workflow of SILAC
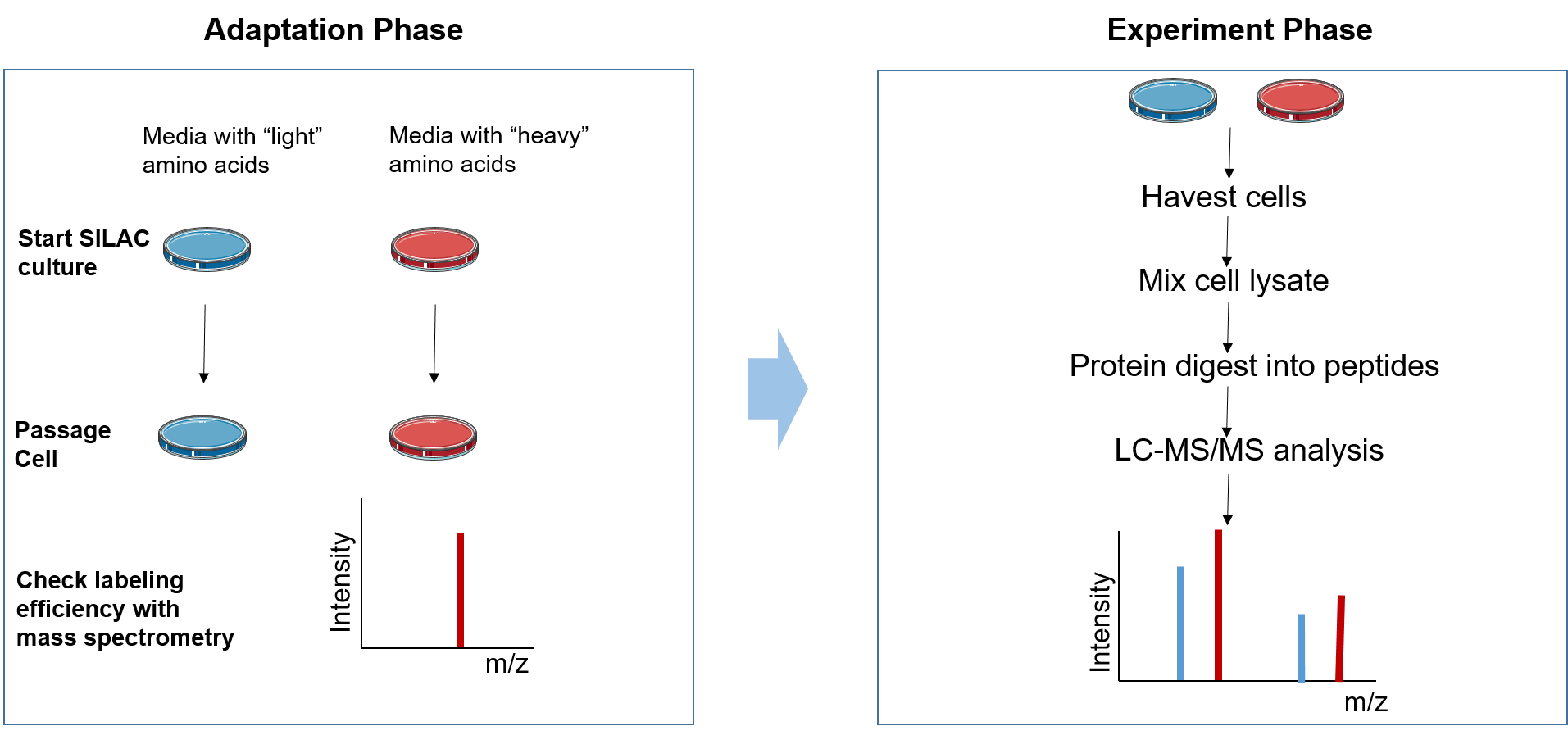
Figure 2. The principle of SILAC
The SILAC experiment can be divided into two phases: the adaptation phase and the experimental phase. In the adaptation phase, cells are grown in light and heavy SILAC medium until heavy amino acids are completely incorporated into the growing cells, which can be assessed by MS. During the experimental phase, the two cell populations are subjected to different treatments according to the research aim. Subsequently, the mixed cell population or protein lysate is studied. Then samples are analyzed with LC-MS/MS to identify and quantify the ratios of heavy peptides to light peptides.
Applications of SILAC
SILAC coupled with LC-MS/MS has been widely used to characterize protein quantitative differences between different samples, investigate the changes of protein post-translational modifications (PTMs), and distinguish specific interacting proteins in the protein-protein interactions (PPIs) networks. There are several applications for SILAC.
- Expression proteomics. SILAC provides an in vivo strategy to label the proteins with different stable isotopic forms of the amino acids which makes it possible to monitor quantitative differences at the protein level under different conditions. In addition, SILAC was also applied to identify differentially expressed proteins in organelles, such as nucleus, nucleolus or-cell insulin secretory granules.
- Dynamic changes of PTMs. For PTMomics analysis, SILAC labeling peptides are subject to a fractionation and an enrichment step to improve identification of PTM-peptides. Combined with MS technologies, SILAC allows for the global and dynamic analysis of PTMs, including phosphorylation, acetylation, glycosylation, ubiquitination, methylation. For example, researchers used SILAC and immobilized metal affinity chromatography (IMAC) for phosphopeptide enrichment to quantify phosphorylation changes of proteins in G-protein-coupled receptor signaling pathways in response to pheromone signals in yeast.
- PPIs study. When studying PPIs, protein complexes are immuno-precipitated from the mixture of SILAC-labeled cell lysates. Combined with SILAC, specifically interacting proteins can be efficiently distinguished from nonspecific background proteins. The abundance of specific interaction partners purified from the bait sample is significantly higher than the one from the control sample, resulting in quantified ratios much higher than 1. In contrast, the abundance of nonspecific background proteins should be comparable from both the bait and the control sample, such that their ratio is close to 1. SILAC-based quantitative proteomics can be used to identify the specifically interaction proteins in investigating exogenous PPIs, endogenous PPIs, or inducible PPIs.
SILAC is a simple and powerful method for quantitative analysis of proteins, characterized by quantitative accuracy and reproducibility. Differentially treated samples can be combined at the level of intact cells or protein, namely at the very first step of the experimental workflow, and can be processed together to minimize experimental error or bias. But it is only appropriate for cell samples, which requires a long time due to cell culture. At Creative Proteomics, we are confident to provide professional and reliable SILAC services and other proteomics quantification services, including:
- iTRAQ-based proteomics analysis service
- TMT-based proteomics analysis service
- SILAC-based proteomics analysis service
- Label-free quantification service
- Semi-quantitative proteomics analysis service
Reference:
- Chen X, Wei S, Ji Y, et al. Quantitative proteomics using SILAC: principles, applications, and developments. Proteomics, 2015, 15(18): 3175-3192.