The measurement of differential protein expression provides a direct and accurate way to detect global changes in cells. Quantitative proteomics holds a significant meaning for discovering diagnostic or prognostic protein markers, detecting new therapeutic targets and understanding the basic biologic processes and mechanisms. With the development of mass spectrometry, we can utilize more reliable and dynamic methods of analyzing differential expression of proteins.
Quantitative proteomics can be divided into relative quantification and absolute quantification. Relative quantification is to study the difference of proteome expression among different situations, of which main methods are stable isotope labeling and label-free quantification. Absolute quantification is to obtain the specific expression level of the protein. The following are some techniques commonly used for relative quantitative proteomics.
1.Metabolic labeling—SILAC
Metabolic labeling needs samples to be cultured in medium enriched stable isotopes. SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture) relies on the metabolic incorporation of a given ‘light” or “heavy” form of the amino acid into the proteins. After a number of cell divisions, the protein is labeled by stable isotope. And the proteins from‘light’ and ‘heavy’ culture can be analyzed together by mass spectrometry. Due to the mass difference, the relative quantification is performed according to the area of these two peptides in mass spectrometry. Amino acids, such as leucine, arginine, lysine and so on, are currently commonly used.
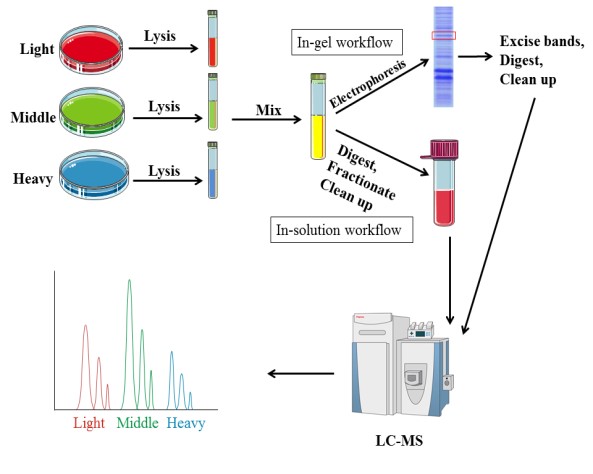
In addition to the research on quantitative protein, SILAC technology is also used in the identification and modification of post-translationally modified proteins, as well as the interaction of proteins. This technology introduces the label before the treatment, thus the error is small and good reproducibility. However, SILAC is only appropriate for cell samples, and it requires a long time due to culturing cells.
2. Chemical labeling—iTRAQ
Chemical label technology introduces isotopes in vitro, which is suitable for cell, body fluids, tissues, and other samples. There are a couple of representative methods, such as isobaric tags for relative and absolute quantitation (iTRAQ) labeling peptide N-terminal amino, esterification label labeling the peptide C-terminal carboxy (-COOH), ICAT for cysteine thiol(-SH)and so on. The following focuses on iTRAQ. iTRAQ is ideally suited for comparing samples in different treatments, biological replicates and provides relative quantitation.
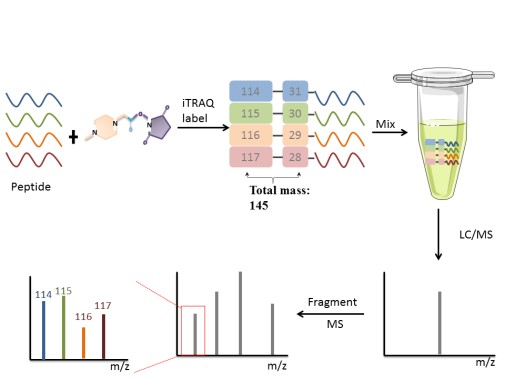
iTRAQ, developed by Applied Biosystems Incorporation, is a technique that utilizes a multiplexed isobaric chemical tagging reagent. The reagent consists of report group, balance group, and peptide reactive group. The peptide reactive group covalently links an iTRAQ Reagent isobaric tag with each lysine side chain and N-terminus group of a peptide, labeling all peptides in a given sample digest. In the MS/MS map, the same protein in different samples labels performance the same mass-to-charge ratio. Quantitation is achieved by comparison of the peak areas and the resultant peak ratios for MS/MS reporter ions.
3.Label-free
Label-free quantificationtechnology can determine the relative amount of proteins in two or more samples. Label-free quantification does not introduce stable isotope to protein, which is different from other methods for protein quantification.
There are often two ways of label-free quantification technology. Quantitative values can be measured through the extraction of the area of the precursor ions’ chromatographic peaks— the area under the curve (AUC) or MS1 signal intensity methods. The second one is based on the number spectra matched assigned to a given protein, called spectral counts. In spectral counts methods, the number of identified MS/MS spectra from the same protein directly correlates with protein abundance. In general, the higher the abundance of a protein in a sample, the more peptides are produced by cleavage. And the more peptides produced by digestion, the more unique matched peptides identified by mass spectrometry and the more corresponding number of spectra.
Proteomics is kind of systematic analysis of all proteins expressed by a cell or tissue, and mass spectrometry is an essential analytical tool. Mass spectrometry can be used to detect changes of the proteome in a cell or tissue that reacts to the exterior or internal disturbances, called quantitative proteomics. Different quantitative analysis methods are selected for different samples and purposes, shown as Table1.
Table 1. Comparison ofdifferent quantification methods
| Method | Sample typle | Variation | Precision | Time |
| SILAC | Cells | Protein purification | High | Long |
| iTRAQ | Any type | Protein purification, digestion | High | Short |
| Label-free | Any type | Protein purification, digestion, separation, MS analysis | Low | Short |
AtCreative Proteomics, we are confident to provide professional and reliable proteomics quantification services, including but not limited to:
- iTRAQ-based proteomics analysis service
- TMT-based proteomics analysis service
- SILAC-based proteomics analysis service
- Label-free quantification service
- Semi-quantitative proteomics analysis service
Reference
1. Deracinois B, Flahaut C, Duban-Deweer S, et al. Comparative and quantitative global proteomics approaches: an overview. Proteomes, 2013, 1(3): 180-218.