
The aim of proteomics is to completely identify and quantify the entire protein samples that we are interested in. And mass spectrometry is one of the most popular methods to study proteomics. There are many different methods in use today to acquire data on a mass spectrometer. Data-dependent acquisition (DDA) mode, also known as Information Dependent Acquisition mode (IDA), is the mode of data collection in tandem mass spectrometry. An alternative toDDA is DIA which is the shortened form of “data independent acquisition”. In DDA mode, all peptides within a certain mass range are fragmented in tandem mass spectrometry.
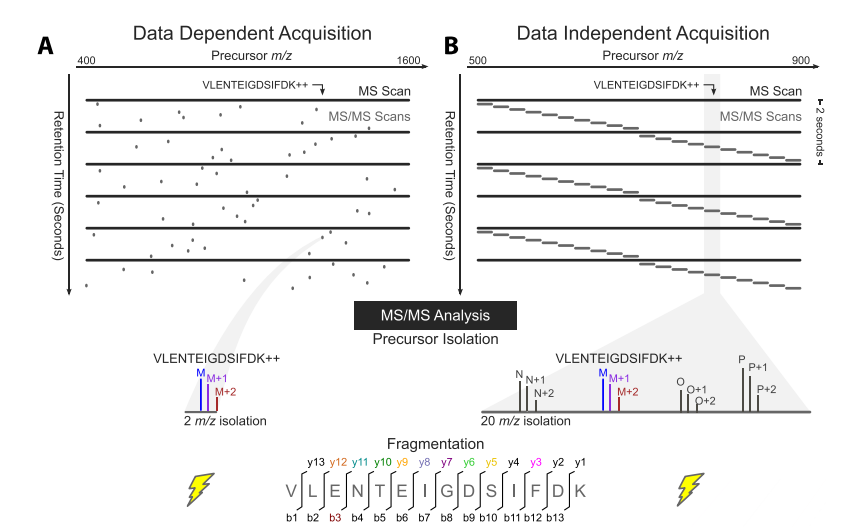
In DDA mode, the mass spectrometer selects the most intense peptide ions in a first stage of tandem mass spectrometry, and then they are fragmented and analyzed in a second stage of tandem mass spectrometry. Otherwise, in DIA mode, for each cycle, the instrument focuses on a narrow mass window of precursors and acquires MS/MSdata from all precursors detected within that window. This mass window is then stepped across the entire mass range, systematically collecting MS/MS data from every mass and from all detected precursors.
The most common method to generate DIA data is called SWATH (SequentialWindowed Acquisition of All Theoretical Fragment ions), in which the mass spectrometer divides the mass range into small mass windows. In SWATH, amass analyzer cover 400-1200 m/z as an overall mass range and the mass spectrometer steps with 2-4seconds cycle time by precursor acquisition windows. During each cycle, the mass spectrometer thus fragments all precursors from the quadrupole isolation window (such as 475 - 500 m/z for 25 wide windows) and a complete, high accuracy fragment ion spectrum of all precursors selected in that isolation window is recorded. The same precursor isolation window is fragmented over and over at each cycle during the entire chromatographic separation, thus providing a time-resolved recording of the fragment ions of all the peptide precursors that elute on the chromatography. The SWATH MS data consists therefore of highly multiplexed fragment ion maps that are deterministically recorded over the user-defined mass precursor mass range and chromatographic separation.
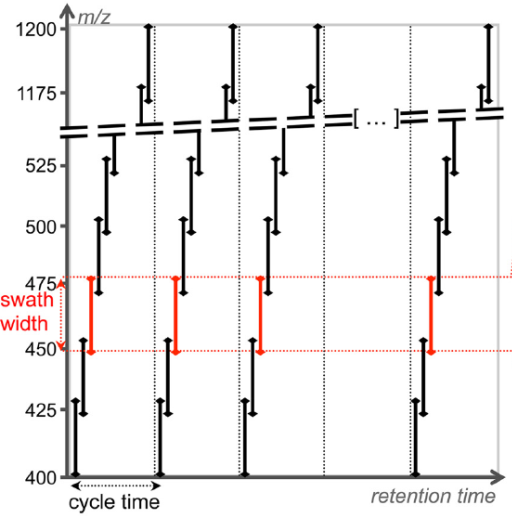
The reasons that the chances of getting interfering compounds are very low and there is no need to know beforehand what will be quantified to make SWATH great. SWATH is not only available for identification but also for quantification. And the quantitation is comparable to MRM which is considered as the gold standard for quantitation through mass spectrometry. The ability to identify and quantify samples in the complex mixture in a large dynamic range and reproducible manner makes it possible to be applied to comparative research studies.
There are still some problems to be solved. For example, the new software tools applied to DIA analysis need to handle some questions, such as how to properly filter false positives and false negatives as well as the deconvolution of complex spectra. However, the future prospect is positive, and various researchers are working on novel bioinformatics techniques to address these issues and increase there producibility, fidelity, and identification breadth of DIA.
References
1. Hu A, Noble W S, Wolf-Yadlin A.Technical advances in proteomics: new developments in data-independent acquisition. F1000Research, 2016, 5.
2. Gillet L C, Navarro P, Tate S, et al.Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Molecular & Cellular Proteomics, 2012, 11(6): O111. 016717.