
Label-free protein quantification is a mass spectrometry-based method for identifying and quantifying relative changes in two or more biological samples instead of using a stable isotope-containing compound to label proteins. There is no limit on the number of samples in label-free protein quantification and it’s in principle applicable to any kind of sample, including materials that can not be directly metabolically labeled (for instance, many clinical samples). It is ideal for large-sample analyses in clinical screening or biomarker discovery experiments.
Principles
In general, label-free approaches can be divided into two distinct groups according to the method used for data extraction. On one hand, the quantification can be inferred by counting the number of peptides or spectra assigned to a given protein, and therefore are generically called spectral counting methods. On the other hand, when liquid chromatography is coupled with mass spectrometry, quantitative values can be measured through the extraction of the area of the precursor ions’ chromatographic peaks— the area under the curve (AUC) or MS1signal intensity methods.
As for spectral counting methods, relative protein quantification is achieved by comparing the numbers of identified MS/MS spectra from the same protein, which has been shown to directly correlate with protein abundance. Some researchers think it is possible because an increase in protein abundance typically results in an increase in the number of its proteolytic peptides. The increasing amounts of digests usually lead to an increase in protein sequence coverage, the number of identified unique peptides, and the number of identified total MS/MS spectra(spectral count) for each protein. Due to ease of implementation, no specific tools or algorithms have been developed specially for spectral counting approaches. But normalization and statistical analysis of spectral counting datasets are necessary for accurate and reliable detection of protein changes in complex mixtures.
As for ion intensity methods, it has been observed that signal intensity from electrospray ionization (ESI) correlates with ion concentration. The height or area of apeak at a particular mass-to-charge ratio (m/z) from a mass spectrum reflects the number of ions for that m/z detected by the mass spectrometer at any given time, which is typically known as the ion abundance. Although the ion abundance cannot be used to directly infer absolute protein or peptide concentration (due to different ionization efficiency for each peptide), comparing the ratio of ion abundances between identical peptides obtained in different experiment runs can be used to estimate differential expression. Some software is available for protein quantification methods based on MS peak intensity of identified peptides, such as Pavel, Drik, MassView, SIEVE, ProteinLynx, Nathan and so on.
Workflow
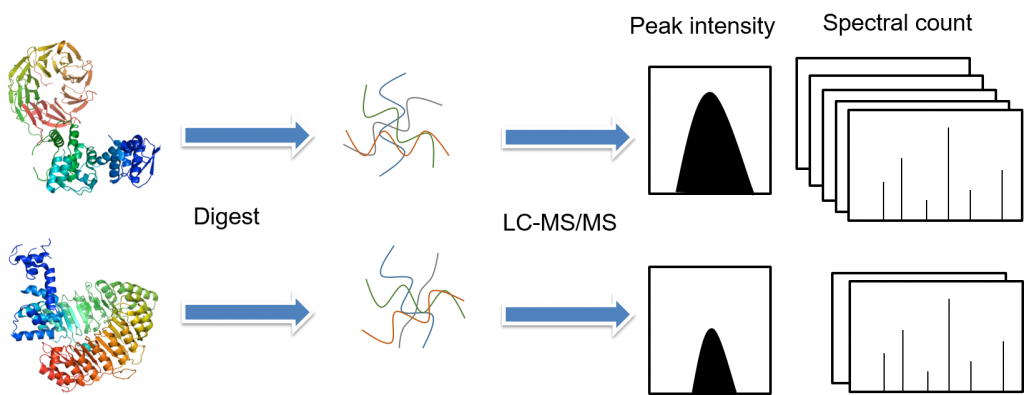
There are several fundamental steps involved in the label-free quantitative proteomics, including sample preparation (protein extraction, reduction, alkylation, and digestion), sample separation by liquid chromatography and analysis by tandem mass spectrometry, and data analysis (peptide/protein identification, quantification, and statistical analysis). In label-free quantitativeproteomics methods, each sample is separately prepared and then subjected toindividual LC-MS/MS or LC/LC-MS/MS runs. Quantification is based on thecomparison of peak intensity of the same peptide or the spectral count of thesame protein.
Advantages and Limitations
Label-free quantitation is an attractive approach for three major reasons. First, the variability that chemical labeling/tagging may introduce is eliminated. Second, chemical and metabolic tags are usually expensive. Third, the time for sample preparation is significantly reduced by the elimination of numerous steps. While the relative abundance of chemical or metabolic tags can be easily measured,label-free quantitation must rely on other parameters such as the peptide or spectral count, which also has inherent limitations. Other issues associated with label-free MS-based quantitation are sequence coverage and the degree of complex sample fractionations prior to analysis in a mass spectrometer. All of these issues and limitations need to be carefully considered before a decision of the optimal approach for a specific experiment.
The label-free quantitative method has been widely used as one of the most commonly used quantitative methods, such as identifying expression profiles in different biological processes, diagnosing certain diseases and cancer biomarkers, monitoring changes in certain biological process proteomes, and studying protein interaction networks, etc. Label-free methods have been widely used for quantitative analysis of proteins. At Creative Proteomics, we are confident to provide professional and reliableiTRAQ services and other proteomics quantification services, including but not limited to:
- ·iTRAQ-based proteomics analysis service
- TMT-based proteomics analysis service
- SILAC-based proteomics analysis service
- Label-free quantification service
- Semi-quantitative proteomics analysis service
References
- Zhu W, Smith J W, Huang C M. Mass spectrometry-based label-free quantitative proteomics. BioMed Research International, 2009, 2010.
- Wong J W H, Cagney G. An overview of label-free quantitation methods in proteomics by mass spectrometry. Proteome bioinformatics, 2010: 273-283.