Background
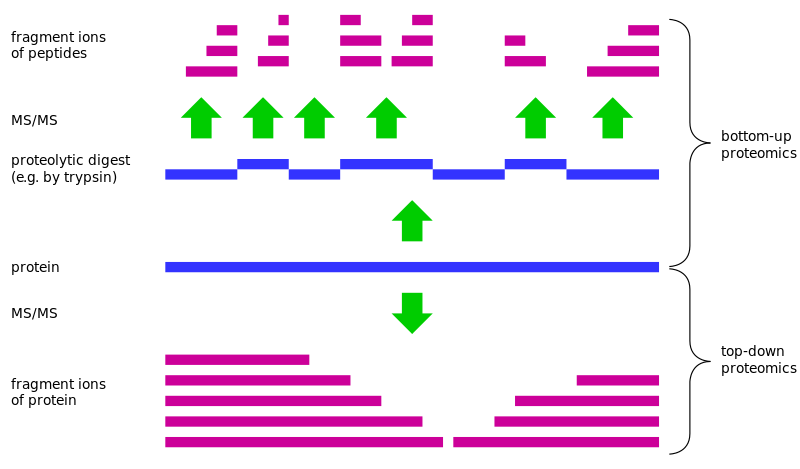
Proteomics is a central issue in biology at the beginning of the 21st century, in which large-scale protein identification is a fundamental point in the calculation of proteomics. Currently, the widely used approach is called bottom-up strategy. The complex sample of the protein is usually digested to produce a mixture of peptides which are separated by liquid chromatography and other techniques, and the peptides are fragmented by mass spectrometry and analyzed according to the Ion peak information to search the database and identify the peptide.Finally, the identified peptides were assembled for protein identification. However, there are about 10% to 20% of the identified protein with this peptide-based method for the identification of the protein sequence. In this case, they often lead to uncertainty in the results of protein reasoning due to the high degree of similarity between the proteoforms in the amino acid sequence.
Brief Introduction for Top-down Proteomics
Top-down Proteomics can take the complete protein as the object of analysis instead of the process of digestion, and it can provide complete protein more accurate and abundant biological information to settle a fundamental solution for the issues existing in Bottom-up proteomics. The "top" refers to the exact determination of the mass of the intact protein, whereas the "down" means the fragmentation of intact proteins by tandem mass spectrometry. Protein sequence coverage rate can reach 100% with the integrity of the protein quality and fragmentation of the information. Comparing with bottom-up proteomics, it can retain a variety of post-translational modification between the relevant information, and gradually become the complementary advantage of bottom-up proteomics.
The Development of Top-down Proteomics
The goal of the human proteome project is to identify all the protein molecules that make up the body so that we can better understand and detect various diseases, and developing a reference list of proteins for drug therapy. With the rapid development of mass spectrometry technology, the number of human protein identification has been refreshed, there are 15646 protein being identified based on the mass spectrometry data, antibodies, amino acid sequencing, 3D structure and other aspects in 20128 protein coding genes, furthermore, there are still 3844 proteins that are lack of experimental evidence except for the 638 dubious. For finding these dubious, scientists need to consider the highly homologous proteins as current mass spectrometry-based identification strategy is mostly based on the digestion, and the identified protein will present as group, that makes it more complex to confirm which protein does exist due to the identified peptides simultaneously matching several highly homologous proteins, and this phenomenon is quite normal in human proteins. Top-down proteomics with complete protein as the object of study provides new ideas and techniques to solve this issue.
In recent years, mass spectrometry techniques that focused on intact proteins analysis are becoming more mature, especially the advancement for mass spectrometry focusing on the core of orbital ionospheric. This shows that mass spectrometry plays a very critical role in Top-down proteomics research.
Complete Mass Spectrometry of Protein
The study of Top-down proteomics mass spectrometry mainly includes two aspects: Using high-resolution mass spectrometry to measure the accurate molecular content of intact proteins; obtaining complete fragmentation of the protein spectrum with high precision tandem mass spectrometry:
1.The exact molecular weight of intact proteins can provide deterministic information for protein identification and post-translational modification analysis which makes it become the primary task in top-down proteomics. ESI and MALDI technology is designed for biological macromolecules, such as protein molecules.
2.The tandem mass spectrometry data contains the amino acid information which can be used to invert the sequence. Therefore, the tandem mass spectrometry data of intact proteins is also the basis for sequence identification, as in the identification of peptide tandem mass spectrometry. Common used technology includes: Collision Induced Dissociation, Electron Capture Dissociation, Fourier Transform Ion Cyclotron Resonance, Electron Transfer Dissociation and High-energy Collision Dissociation etc.
The Challenge and Opportunities
At present, the biggest challenge for top-down proteomics is not separation technology or mass spectrometry, but bioinformatics - the algorithm and software for mass spectrometry data analysis. The top-down based on intact protein and the bottom-up based on the digested peptide form a complementary proteome technology system, both of which have advantages and applicable problems. After a large number of accurate identification of the proteoforms, scientists are committed to studying about another important research topics - the quantitative analysis of proteoforms.
Related Service: