Role of Disulfide Bonds in Protein Folding & Stability
Disulfide bonds act as intramolecular bridges. They impose constraints on the polypeptide chain. This reduces conformational entropy. As a result, the protein attains its native fold more rapidly. Furthermore, disulfide cross-links enhance resilience against thermal and chemical denaturation. These bonds are indispensable for functional integrity in extracellular proteins and secreted enzymes.
Disulfide bonds also modulate protein dynamics. They can stabilize specific domains. In some cases, they trigger conformational switches. These switches are critical in redox-regulated signalling pathways.
Mass Spectrometry Approaches for Disulfide Mapping
Mass spectrometry (MS) is a central analytical technique for the identification and localization of disulfide bonds in proteins. Its high sensitivity, resolution, and capacity for structural elucidation enable precise mapping of disulfide connectivity in both simple and complex protein systems.
Bottom-Up Proteomics Approaches
In bottom-up proteomics, the protein of interest is enzymatically digested into smaller peptide fragments, which are then subjected to liquid chromatography-mass spectrometry (LC-MS) analysis. For disulfide bond mapping, non-reducing digestion conditions are employed to preserve native disulfide linkages between cysteine residues. As a result, disulfide-linked peptides are retained as covalently connected species.
Identifying disulfide-linked peptides relies on observing precursor ions with higher molecular weights and the presence of unique fragmentation patterns during tandem mass spectrometry (MS/MS). Common fragmentation techniques include collision-induced dissociation (CID), higher-energy collisional dissociation (HCD), and electron transfer dissociation (ETD), with ETD being particularly useful for preserving labile disulfide linkages during analysis.
The workflow often includes parallel analyses under reduced and non-reduced conditions. In reduced samples, disulfide bonds are cleaved using agents such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP), followed by alkylation of free thiol groups with reagents like iodoacetamide (IAM) to prevent reoxidation. Comparing reduced and non-reduced spectra facilitates the unambiguous assignment of disulfide-linked peptides and confirmation of cysteine connectivity.
Top-Down and Middle-Down Analysis
Top-down proteomics involves the direct analysis of intact proteins without prior enzymatic digestion. This method preserves all native post-translational modifications (PTMs), including disulfide bonds, allowing for the observation of complete disulfide bonding patterns in the full protein context. High-resolution MS platforms, such as Fourier-transform ion cyclotron resonance (FT-ICR) and Orbitrap instruments, are required due to the complexity of intact protein spectra. The top-down analysis detects disulfide scrambling, isoform variations, and higher-order structural features. However, the technique is technically demanding and often limited to small or medium-sized proteins.
Middle-down proteomics provides a compromise between top-down and bottom-up approaches. Partial proteolysis generates larger peptide fragments that retain more structural information than typical tryptic peptides. These fragments are particularly advantageous for studying disulfide-rich proteins, as they often preserve disulfide linkages that are lost in conventional bottom-up workflows. Fragmentation methods like ETD or electron capture dissociation (ECD) are essential for analyzing these larger peptides with intact disulfide bonds.
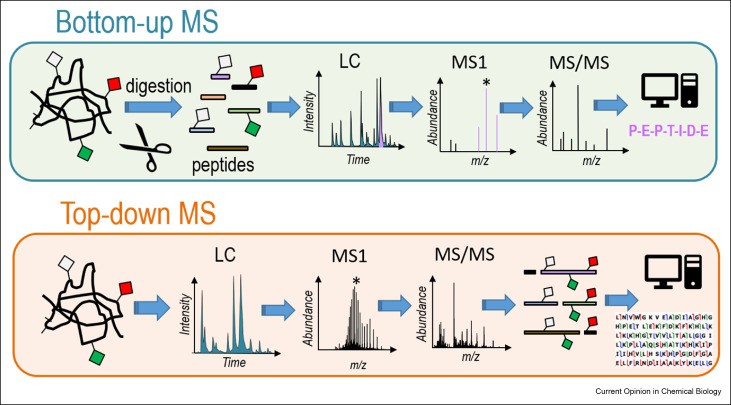 Figure 1. Schematic representation of bottom-up and top-down strategies. (Brodbelt J S, 2022)
Figure 1. Schematic representation of bottom-up and top-down strategies. (Brodbelt J S, 2022)
Enzymatic Digestion Protocols for Disulfide Peptides
Enzymatic digestion plays a critical role in the disulfide mapping workflow. Proteases must be chosen based on specificity, cleavage pattern, and compatibility with disulfide bond preservation. Trypsin is the most commonly used protease, cleaving at the C-terminal side of lysine and arginine residues. However, its activity can be hindered when disulfide bonds constrain access to cleavage sites.
Alternative proteases such as Glu-C, Asp-N, chymotrypsin, or thermolysin can generate complementary peptide fragments. Using multiple enzymes in sequential or parallel digestions enhances the coverage and resolution of disulfide bond mapping.
Digestion must be performed under non-reducing and non-denaturing conditions to preserve disulfide linkages. Denaturants such as urea or guanidine hydrochloride may be used at low concentrations to improve solubilization without disrupting disulfide integrity. Partial reduction strategies, in which a limited amount of reducing agent is applied, can be employed to cleave selected disulfide bonds while leaving others intact. This technique allows sequential mapping of disulfide networks in multi-cysteine proteins.
Chromatographic Techniques for Disulfide Separation
Chromatographic separation is frequently coupled with MS to resolve disulfide-linked peptides and improve analytical clarity.
HPLC Methods for Disulfide-Linked Peptides
Reversed-phase HPLC employs hydrophobic stationary phases, typically C18, to separate peptides by nonpolar surface area. Disulfide-bridged peptides exhibit increased hydrophobicity relative to their reduced counterparts. Gradient elution, using water and acetonitrile with 0.1% formic acid, resolves these species. Steeper gradients accelerate elution but may compromise peak capacity. Shallow gradients yield sharper peaks. Column temperature, flow rate, and particle size are optimized to balance sensitivity and throughput. Ultra-high-pressure liquid chromatography (UHPLC) further enhances resolution by using sub-2 µm particles and elevated pressures.
Ion Exchange & Size-Exclusion Chromatography Applications
Strong-cation exchange chromatography separates peptides by net positive charge. Disulfide formation can alter peptide pI and retention time. Stepwise salt gradients elute bound species selectively. Anion exchange may be used when peptides carry negative charges. Size-exclusion chromatography fractionates molecules by hydrodynamic volume. Disulfide-linked dimers or higher-order aggregates elute earlier than monomeric peptides. SEC buffers are chosen to maintain native structure and minimize non-specific interactions. Combining SEC with online MS ensures direct analysis of each size-fractionated component.
Spectroscopic Assays for Quantifying Disulfide Bonds
Spectroscopic techniques provide quantitative and qualitative data on disulfide content in proteins.
Ellman's Reagent Titration Assays
Ellman's reagent (DTNB) reacts stoichiometrically with free thiol groups to produce 2-nitro-5-thiobenzoate (TNB²⁻), a yellow chromophore. The absorbance maximum at 412 nm correlates directly with thiol concentration. First, the native protein sample is assayed to establish baseline free thiols. Next, a reducing agent (e.g., dithiothreitol) liberates all disulfide-linked thiols, and DTNB is added. The increase in absorbance quantifies total cysteine residues. The difference between total and native thiols yields the number of disulfide bonds. This assay is sensitive to low micromolar concentrations and requires minimal sample preparation. Buffer components must be free of thiol-reactive contaminants. Assay pH is typically maintained at 7.5 to 8.0 to optimize reaction kinetics..
Circular Dichroism: Monitoring Folding & Bond Formation
CD spectroscopy measures the differential absorption of left- and right-circularly polarized light by chiral chromophores. CD signals in the far-UV region (190–250 nm) reflect peptide backbone conformation. Formation or reduction of disulfide bonds induces subtle spectral shifts. Specifically, disulfide bond formation often increases α-helical content, leading to more pronounced negative ellipticity at 208 and 222 nm. Kinetic experiments can monitor folding pathways by recording time-resolved CD spectra following oxidative folding conditions. Thermal unfolding assays further assess the stabilizing effect of disulfide bonds by measuring ellipticity changes as a temperature function. Data fitting yields melting temperatures (T_m), providing insight into bond-induced stability. CD assays require protein concentrations of 0.1–1 mg/mL and buffer systems transparent in the far-UV range.
Advanced & Emerging Technologies
Recent innovations have expanded the analytical repertoire for disulfide bond characterization. High-resolution nuclear magnetic resonance (NMR) spectroscopy now permits direct observation of disulfide linkages within small to medium-sized proteins. Modern NMR probes achieve sub-ppm sensitivity. They detect characteristic cysteine Cβ chemical shifts and inter-residue nuclear Overhauser effects. This capability enables precise localization of each covalent sulfur–sulfur bond in solution. Cryogenic electron microscopy (cryo-EM), now capable of near-atomic resolution, enables visualization of disulfide-stabilized domains in large complexes. It is especially valuable in cases where crystallography is not feasible. In silico prediction tools, using machine learning and structural databases, model potential disulfide connectivity based on sequence and structural context. These tools can guide experimental design and identify aberrant disulfide formations.
Disulfide Bond Analysis in Biopharmaceuticals
Monoclonal Antibody Disulfide Characterization
Monoclonal antibodies (mAbs) possess a defined network of inter- and intra-chain disulfide bonds that underpin quaternary structure and antigen affinity. A typical human IgG1 contains twelve cysteine residues forming four inter-chain and two intra-chain disulfides per heavy-light pair. Analytical workflows typically involve non-reducing enzymatic digestion followed by mass spectrometry and chromatographic separation to identify native disulfide linkages and detect aberrant scrambling or bond cleavage. Ensuring correct disulfide pairing directly correlates with therapeutic efficacy and immunogenicity risk reduction.
Recombinant Protein Therapeutics Quality Control
Recombinant proteins often display heterogeneous disulfide profiles due to oxidative folding in expression hosts. For insulin analogs, the correct pairing of A- and B-chain disulfides is essential for bioactivity. Analytical workflows integrate size-exclusion chromatography to separate mispaired isoforms, followed by MS-based technology using Glu-C digestion to generate peptides containing single disulfide bridges. Quantitative comparison of extracted ion chromatogram peak areas yields a relative abundance of correctly folded versus aberrant species, supporting process optimization.
Comparative Disulfide Mapping of Mutant vs. Wild-Type Proteins
Site-directed mutants altering cysteine residues can disrupt native disulfide geometry. In tumor necrosis factor receptor mutants, loss of intra-domain disulfide yields misfolded aggregates. Top-down MS under non-reducing conditions reveals mass shifts corresponding to missing bonds. Complementary hydrogen-deuterium exchange mass spectrometry detects enhanced solvent accessibility in the mutant's loop regions. Such comparative studies guide protein engineering by correlating specific disulfide linkages with structural stability and function.
Case of Disulfide Bond Analysis
Disulfide bond characterization of endogenous IgG3 monoclonal antibodies using LC-MS: an investigation of IgG3 disulfide-mediated isoforms
Journal: Analytical Methods
Published: 2016
DOI: 10.1039/C6AY01248E
Background
Monoclonal antibodies (mAbs) rely on correct disulfide bond connectivity for proper folding, stability, and biological activity. While classical disulfide linkages in all four human IgG subclasses have been known since the 1960s, alternative, disulfide-mediated isoforms have been observed in IgG2, IgG4, and, to a lesser extent, IgG1. IgG3, despite its potent effector functions, has not been similarly investigated, likely owing to its historically short serum half-life and lack of therapeutic development.
Purpose
To determine whether endogenous human IgG3 exhibits alternative disulfide bonds in its constant region—analogous to the isoforms seen in other IgG subclasses—by rigorously mapping all native disulfide linkages. Establishing the absence or presence of such isoforms informs the design and production of recombinant IgG3-based therapeutics.
Methods
- Sample Preparation: Native IgG3 was isolated from human serum gamma-globulins via sequential Protein A (to deplete IgG1/2/4) and Protein G affinity chromatography, then validated by SDS-PAGE and Western blot.
- Proteolysis under Non-Reducing Conditions: Samples were denatured, free cysteines were capped with N-ethylmaleimide, and trypsin was digested.
- LC-MS/MS Analysis: Tryptic peptides were separated by reversed-phase HPLC and analyzed on an Orbitrap Velos Pro with both Electron Transfer Dissociation (ETD) and Collision-Induced Dissociation (CID).
- XIC/ETD Approach: Extracted ion chromatograms of Cys-containing peptides were compared to detect co-eluting pairs. ETD spectra were then interrogated for marker ions and c/z fragments to confirm specific disulfide bonds.
- CID Confirmation: For disulfide bonds that eluded ETD assignment (Hinge-2, Hinge-3, CH1), intact dipeptide masses were first located in high-resolution MS data, then CID spectra were used to verify peptide fragmentation patterns.
Results
- Classical Disulfide Bonds: All 21 constant-region disulfide bonds—covering CH1, CH2, CH3, and inter- and intra-chain linkages for both κ and λ light chains—were unambiguously assigned using the combined XIC/ETD and CID strategies.
- Identical-Peptide Dimers: The methodology was refined to distinguish identical hinge-region dipeptides by comparing XICs of different charge states, confirming all four expected inter-chain hinge bonds.
- Absence of Alternative Isoforms: No evidence was found for any non-classical disulfide linkages in endogenous IgG3. Extensive scrutiny of all Cys-containing peptides across two independent IgG3 batches and multiple runs failed to detect any aberrant pairing.
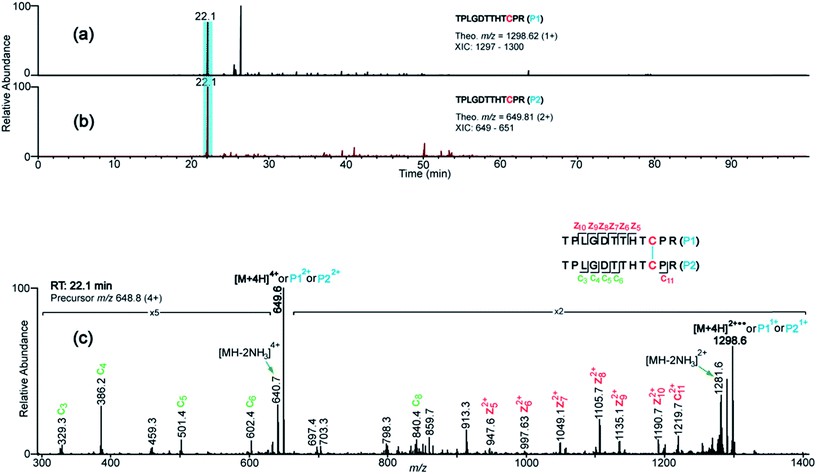 Figure 2. XIC's and ETD spectrum showing assignment of the CH2 domain disulfide bond.
Figure 2. XIC's and ETD spectrum showing assignment of the CH2 domain disulfide bond.
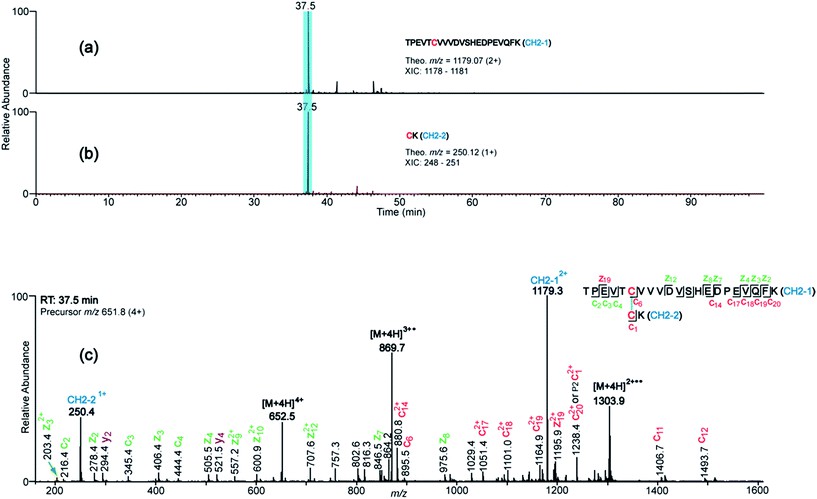 Figure 3. Representative XIC's (a and b) and ETD spectrum (c) that support the assignment of the Hinge-1 disulfide, which has identical Cys-containing peptides.
Figure 3. Representative XIC's (a and b) and ETD spectrum (c) that support the assignment of the Hinge-1 disulfide, which has identical Cys-containing peptides.
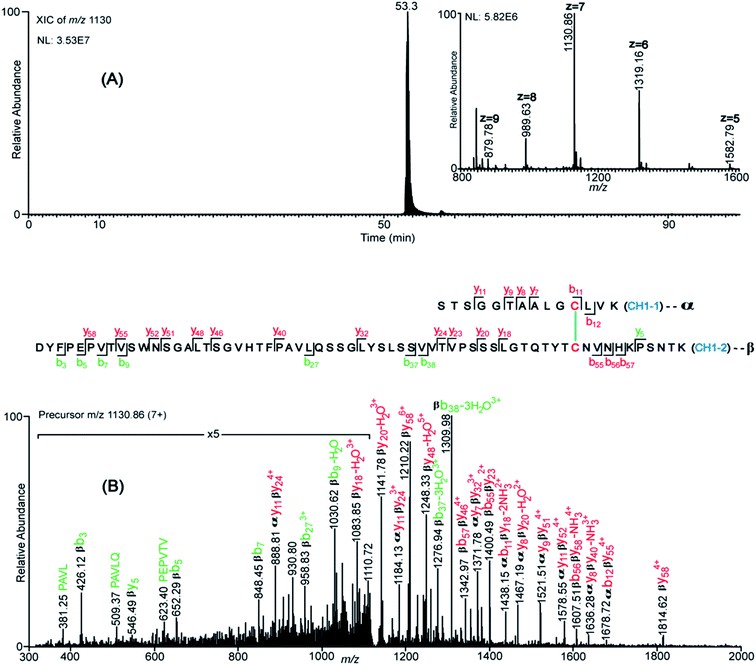 Figure 4. XIC and CID spectrum supporting the assignment of the CH1 domain disulfide bond. (A) XIC of the CH1 domain tryptic dipeptide at m/z 1130 (7+).
Figure 4. XIC and CID spectrum supporting the assignment of the CH1 domain disulfide bond. (A) XIC of the CH1 domain tryptic dipeptide at m/z 1130 (7+).
Conclusion
Endogenous human IgG3 strictly adheres to the classical disulfide bonding pattern in its constant region, with no alternative disulfide-mediated isoforms detectable by advanced LC-MS/MS techniques. This finding—the first of its kind for IgG3—implies that recombinant IgG3 therapeutics need only reproduce the canonical disulfide framework to ensure proper structure and function.
References
- Brodbelt J S. Deciphering combinatorial post-translational modifications by top-down mass spectrometry. Current opinion in chemical biology, 2022, 70: 102180. DOI: 10.1016/j.cbpa.2022.102180
- Tholey A, Becker A. Top-down proteomics for the analysis of proteolytic events-Methods, applications and perspectives. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 2017, 1864(11): 2191-2199. DOI: 10.1016/j.bbamcr.2017.07.002
Related Articals
Our products and services are for research use only.
