
Abstract
Mass spectrometry has become an important technology to connect proteins and genes, laying the foundation for large-scale automated protein identification. There are two major parts of the mass spectrometry available for proteins or peptides analysis.One is the MALDI-TOF technology, as a pulsed ionization technique, it generating ions from solid-phase specimens and measuring their molecular weight in flight tubes, the other is ESI-MS, which is a continuous ionization method that generates ions from the liquid phase and measures their molecular weight in conjunction with quadrupole mass spectrometry or in a time-of-flight detector. Mass spectrometry devices and technologies have made great progress in recent years.
In MALDI-TOF, the most important advances are ion reflectrons and delayed ion extraction, which can achieve fairly accurate molecular weights. In ESI-MS, the presence of a nano-electrospray source makes it possible to analyze microscaled samples within 30 to 40 minutes.
The combination of reversed-phase liquid chromatography and tandem mass spectrometry (tandem MS) enables the detection of dozens of picomole levels. If capillary chromatography is used in conjunction with tandem mass spectrometry, it can be detected at low levels of picomole to high femtomole levels; while utilizing capillary electrophoresis in conjunction with tandem mass spectrometry, it can be detected at levels less than femtomole, even can be processed at attomole level.
At present, most of the proteins are identified by the combination of enzymatic hydrolysis, liquid chromatography, tandem mass spectrometry, and computer algorithms. We will illustrate how to identify proteins by mass spectrometry with peptide mass fingerprinting and sequencing of peptide fragments as follows:
1.Peptide Mass Fingerprinting (PMF)
PMF was first put forward by Henzel et al. in 1993. The protein isolated from 2-DE is cleaved at the C-terminus of arginine or lysine on the gel or on the membrane with an enzyme (most commonly trypsin). The exact molecular weight produced by the fragmentation is measured by mass spectrometry (MALDI-TOF-MS, or ESI-MS), and the mass of peptides that achieved by this technology can be accurate to 0.1 molecular weight. The mass of all the peptides is finally matched to the mass of the theoretical peptides in the database (The theoretical peptides are produced by the enzymes used to break the protein). The matching results are arranged according to the number of peptide fragments shared by the peptide fragment in the database and the unknown protein, and the peptide fragment ranked first may represent an unknown protein. If there is a big difference between the peptide fragments between the runner-up and the runner, and the protein can be covered with the peptide fragments shown in the experiment which indicates the higher possibility of correct identification.
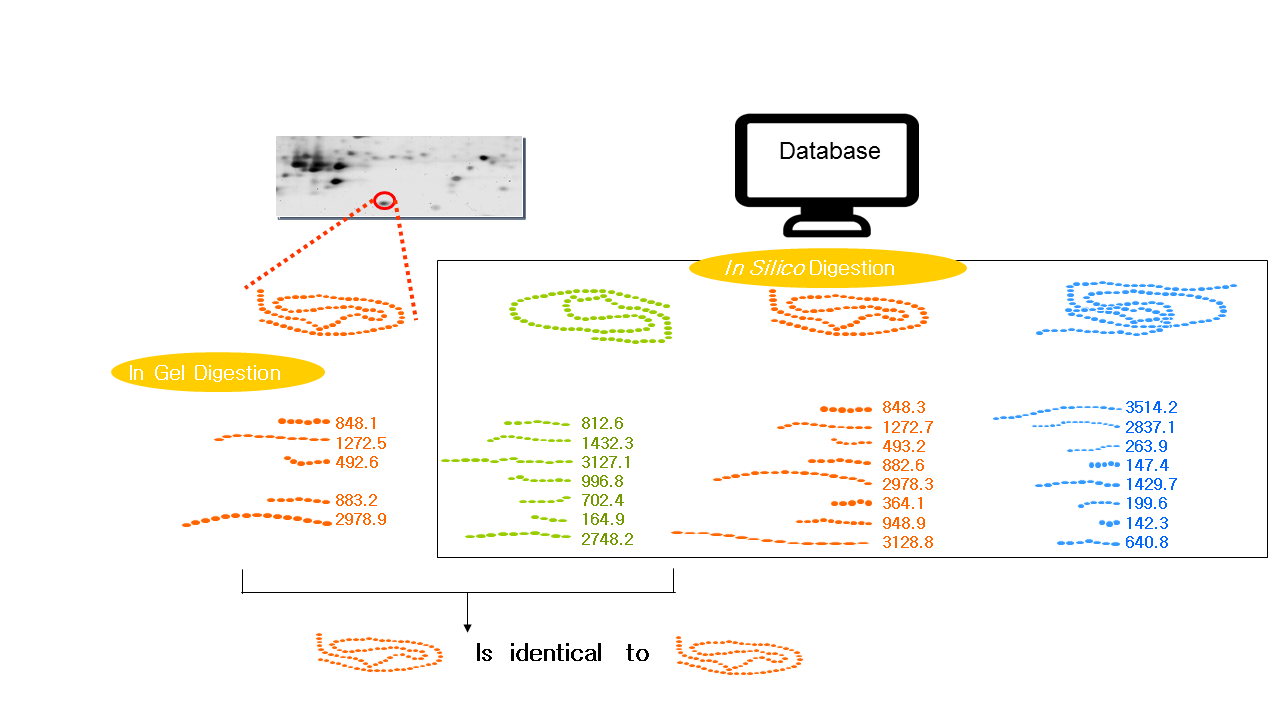
2.Partial sequencing of peptide fragments
peptide mass fingerprinting, on its own, can not reveal the derived peptide fragments or proteins. In order to further identify proteins, a series of mass spectrometry methods have been used to describe peptide fragments. The amino acids are removed sequentially from the N- or C-terminus to form ladder peptides with enzymes or chemical methods. One of the methods is to degrade from the N-terminus in a controlled chemical pattern, and a series of ladder peptide fragments of different sizes can be generated. The resulting amount of peptide mass is measured by MALDI-TOF-MS. Another method involves the application of carboxypeptidases, removing different numbers of amino acids from the C-terminus to form peptide fragments. Chemical and enzymatic methods produce relatively long sequences with molecular weights that are accurate to distinguish between lysine (128.09) and glutamine (128.06). Alternatively, post-source decay (PSD) and collision-induced dissociation (CID) are applied within the mass spectrometer in order to generate a mass spectrum containing a series of peptide peaks that are only different in mass from one amino acid residue. Therefore, the inference of the peptide fragment sequence will be allowed. The analysis of the peptide fragment PSD can generate partial sequence information in the MALDI reactor.
Peptide mass fingerprinting is first performed. Afterward, a meaningful peptide fragment is selected as parent ion in the mass spectrometer and degraded to daughter ion during flight to the ion reactor. In the reactor, different size fragments can be measured to the detector with varying voltages, however, incomplete fragments are often produced. The current method for sequencing with peptide fragments begins with the CID at the end of the 1970s and can be accomplished in a triple quadrupole mass spectrometer ESI-MS or MALDI-TOF-MS combined impactor. In ESI-MS, the peptide ions generated by the electrospray source are measured in the first quadrupole mass spectrometer, and the meaningful peptide fragments are sent to the second quadrupole mass spectrometer, then bombarded into fragments by inert gas. The resulting product is measured in a third quadrupole mass spectrometer. Compared to MALDI-PSD, CID is stable, robust, and prevalent. Peptide ion fragments are bombarded along the backbone of the amide bond to produce trapezoidal sequences. Consecutive fragment differences determine the amino acid mass of that sequence at that point to speculate the sequence. The residues of several sequences that can also be obtained from the CID, are known as peptide sequence tags. Thus, the molecular weight of the combined peptide fragment precursor ion and the distance of the peptide fragment from the N-C terminal will be sufficient to identify a protein.