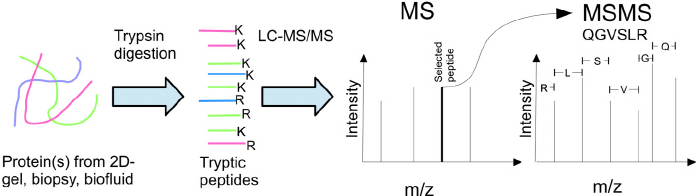
Mass spectrometry plays an important role in protein identification and other experiments. However, the current application is not particularly extensive. Here, we collect some common questions for mass spectrometry identification, hope it will be helpful for you to get a better understanding of it:
Q1: What is the difference between a primary mass spectrometer and a secondary mass spectrometer? And how to choose the proper one?
A: The primary means of mass spectrometry identification mainly refers to the peptide mass fingerprinting(PMF), that is, the application of mass spectrometry to accurately measure the molecular weight of the enzymatic fragments and to compare the library for the identification of proteins, the sequence of the peptide was identified and the results of PMF were combined to achieve the identification of the protein. Secondary mass spectrometry can obtain short sequences of partial peptides with higher reliability.With the increasing stringency of data requirements in current journals, secondary mass spectrometry identification is a major trend in protein identification, and even if a mass spectrometry identification is currently performed, it is necessary to select some PMF results for secondary mass spectrometry validation.
Q2: What should be done if the species being studied is not a model organism?
A: The successful identification of proteins can be achieved by referring to the closest model organisms of genetic relationship. If the mass spectrogram is great but no identification results, it indicates that this may bea brand new protein. De-novo and other technologies can be used for in-depth analysis and identification.
Q3: How to evaluate the effectiveness of mass spectrometry?
A: A PMF score of more than 60 points (P<0.05) is generally considered to be successful. For tandem mass spectrometry, the score exceeds 60 points or although it does not exceed 60 points, there is at least one peptide with a score of more than 30 points will be successfully identified.
Q4: What are the uses of some special mass spectrometry methods?
A: Other uses for mass spectrometry include phosphorylation site analysis, protein sequencing, mixed protein identification (shotgun technology), accurate determination of molecular weight, disulfide bond position analysis, etc.
Q5: Which kind of staining method is better?
A: Coomassie brilliant blue staining is preferred. Silver staining is also acceptable, but the identification success rate is slightly lower, and the use of tandem mass spectrometry for the identification of silver stained proteins can greatly improve the identification success rate.
Q6: Can the gel be preserved for a long period of time? How to maintain? Will it affect the identification effect?
A: If kept the gel for more than one month, it should be wrapped in a plastic wrap and kept in the refrigerator. If the time is less than one month, it can be kept at room temperature without affecting the mass spectrometry.
Q7: How to take the gel point of mass spectrometry? Any precautions?
A: When taking the point, cut the tip of the 0.2ml tip and remove the gel with a pipette tip and transfer it to a 0.5ml centrifuge tube. Rinse the beads two or three times with water and then blot the tube. Keep all moisture seals in place and mark them. For centrifuge tube, it will be better to choose imported centrifuge tube, avoiding plastic pollution. It is best to use deionized water or double distilled water. Take a mask and gloves to prevent keratin contamination.
Q8: Can the gel be stored for a long period? How to store and ship?
A: Protein gels can be stored for long periods of time without affecting mass spectrometry identification. In general, if it is within a week or two, it is better to wrap it in plastic wrap and store it in a 4 degree refrigerator. If the storage time exceeds one month, the protein spots can be removed and placed in a -20℃ degree or -80℃ degree refrigerator without affecting the subsequent mass spectrometry identification. When transporting glue points, it can be transported at normal temperature, and there will not be much impact within three to five days.
Q9: How to choose between primary and secondary mass spectrometry?
A: The primary mass spectrometry identification method is often referred to as peptide mass fingerprinting (PMF). The secondary mass spectrometry identification method is often referred to as tandem mass spectrometry. Early protein identification, considering the cost, the identification of proteins for sequencing organisms can generally be achieved with a primary mass spectrometer, while non-sequencing organisms often choose secondary mass spectrometry. But now, the price of the secondary mass spectrometer is also greatly reduced, not much different from that of the primary mass spectrometer, and both the reliability and the identification success rate are greatly improved.
Q10: What is the general process of mass spectrometry, and what data can be provided by companies?
A: Mass spectrometry identification mainly includes three steps: proteolysis, mass spectrometry data acquisition, and library search. For the identification of each protein spot, three types of data are generally provided, including: mass spectrogram (PDF format, including 1 piece of primarymass spectrum and 5-10 pieces of secondary mass spectra), mass spectrum peak list (text format, mainly the information of some MS fragments) and the search results (general PDF format or excel format for self-built library for localized search; general webpage format offered for Mascot online search).
Q11: What are the general factors that influence the results of mass spectrometry?
A: The unsuccessful mass spectrometry identification can be mainly divided into two categories. One reason is that the mass spectrometry is not effective, and the other is a protein database that has no reference data. The reasons that will lead to the unsuccessful mass spectrometry results can be subdivided as follows: (1) the protein spots are so light that the protein content is too low; (2) the protein spots are too small which is not conducive to operation; (3) Although separated by two-dimensional electrophoresis, the point is a mixture of several proteins; (4) proteolysis and mass spectrometry errors.
To avoid these factors, it is necessary to take take darker spots of protein, and the protein spot area needs to be appropriately larger (for some small but very clear protein spots, the tip of the sample should be selected with a large bore diameter. It does not matter if the blanks on the edges are properly picked up). Avoid point-mixing protein spots and handle rich experience in enzymatic digestion and mass spectrometry. The poor identification results causing by no reference database can be solved by replacing the larger database, taking the EST database, and building a local mass spectrum database based on the protein database of a single species.
Q12: What is the difference between tandem mass spectrometry and mass spectrometry sequencing?
A: Mass spectrometry identification often uses software to automatically match existing databases in order to obtain results. While mass spectrometry sequencing requires direct calculation of the peptide sequence based on the difference in molecular weights of adjacent or similar peaks in the mass spectrogram.
Q13: Whether the unknown protein sequenced by mass spectrometry or protein sequencer?
A: The simpler method will be mass spectrometry, and the cost is not expensive. If the mass spectrometry cannot be determined, it can also be combined with N-terminal sequencing. This two data combination analysis can basically determine the target protein.
All the mentioned above are some issues you may meet while using mass spectrometry. We will keep updating more related and useful information for you.
Related service: