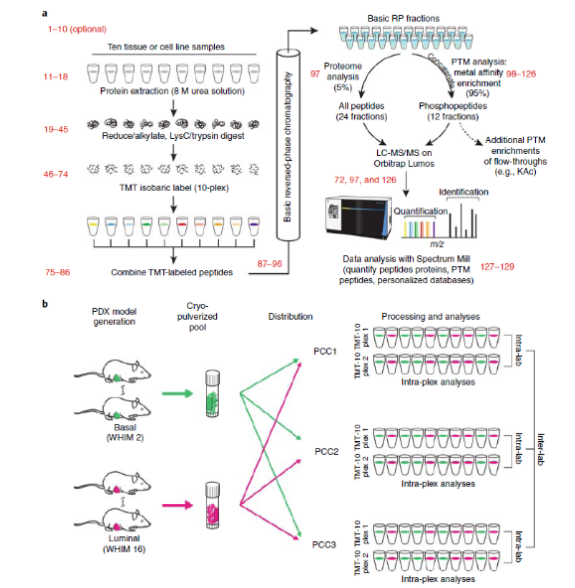
In the past decade, genomics research has systematically mapped genetic changes in human cancer, however, we still know little about the direct effects of these changes on the proteome. Proteomics studies of the NIH Clinical Proteomic Tumor Analysis Consortium (CPTAC) have shown that the integration of proteomic and phosphorylated proteome data with genomic data can improve the identification of tumor pathways.
'We introduced an optimized process that utilizes a 10-label tandem mass tag (TMT) for multiplex analysis and relative quantification so as to enable an overall proteome and phosphorylated proteome analysis of the tissue or cell line,' the authors write in the article.
The researchers also claimed that, 'In the 10,000 proteins quantified per sample, we can distinguish 7,700 human proteins derived from tumor cells, and 3,100 mouse proteins derived from the surrounding matrix and blood.'
The researchers point out that this improved approach has significantly accelerated research compared to past research which took 9 months to complete the quantification and analysis of the proteome and phosphorylated proteome of 100 samples. They also concluded that, 'At present, high quality, deep and repeatable data from within and between laboratories will help integrate mass spectrometry-based proteomics analysis with proteomic-genomics data, thereby offering new biological insights.'
1.Mertins P, Tang L C, Krug K, et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nature protocols, 2018: 1.