What Is Protein Lipidation?
Protein lipidation constitutes a fundamental post-translational modification (PTM) in which lipid moieties covalently attach to specific amino acid residues on proteins. This biochemical process alters the physicochemical properties of proteins, including their membrane affinity, subcellular localization, stability, and interaction networks. Lipidation facilitates the anchoring of proteins to cellular membranes and modulates signaling pathways critical to cellular homeostasis.
The covalent linkage between lipid groups and protein substrates is often enzymatically catalyzed. Protein lipidation is indispensable for numerous biological processes, such as intracellular trafficking, signal transduction, and membrane dynamics. Aberrant lipidation patterns are implicated in pathologies ranging from cancer to neurodegenerative diseases.
Major Types of Protein Lipidation
Several distinct lipidation forms exist, each defined by the nature of the lipid attachment and the amino acid residue involved:
- N-myristoylation: This is when a fatty acid with 14 carbon atoms, called a myristoyl group, attaches to the beginning (N-terminus) of a protein, specifically to a glycine amino acid. This bond is stable and permanent. It helps guide the protein to the cell membrane, where it often carries out its functions.
- S-palmitoylation: A 16-carbon fatty acid known as a palmitoyl group attaches to a cysteine amino acid through a flexible chemical bond. Unlike myristoylation, this modification can be reversed. That means the cell can use it to control where the protein goes and what it does.
- Prenylation (Isoprenylation): Small hydrocarbon groups—such as farnesyl (15 carbon atoms) or geranylgeranyl (20 carbon atoms)—are added to a cysteine near the protein's tail end. This helps the protein stick to membranes and interact with other proteins.
- Glycosylphosphatidylinositol (GPI) Anchoring: This involves attaching a sugar-fat structure (called a GPI anchor) to the end of a protein. It secures the protein to the outer surface of the cell membrane.
- Cholesterylation: This is the addition of a cholesterol molecule to a protein. Although less common, it plays a key role in certain signaling pathways, especially during development.
Select Service
Learn More
Experimental Techniques to Analyse Protein Lipidation
Mass Spectrometry-Based Lipidomics
Mass spectrometry (MS) has become the cornerstone for analyzing protein lipidation. It allows for precise identification of lipid species and their attachment sites at the peptide level.
- Bottom-Up Proteomics
In this approach, proteins are digested enzymatically, typically with trypsin or chymotrypsin. The resulting peptides, including lipid-modified ones, are analyzed by liquid chromatography coupled to tandem MS (LC-MS/MS). Electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI) are frequently employed.
Lipidated peptides show unique mass shifts and fragmentation patterns. Database searching with specialized algorithms (e.g., Sequest, Mascot, MaxQuant) helps identify lipidation sites.
Advantages: High-resolution, site-specific identification
Limitations: Lipid-peptide instability, low abundance, ion suppression
- Top-Down Proteomics
Top-down proteomics analyses intact lipidated proteins without enzymatic digestion. This allows detection of multiple modifications on a single protein molecule. Fourier transform ion cyclotron resonance (FT-ICR) or Orbitrap platforms provide the resolution required.
Top-down MS is particularly valuable when studying proteins with complex or multiple lipid attachments. However, it is technically demanding and often limited to purified proteins.
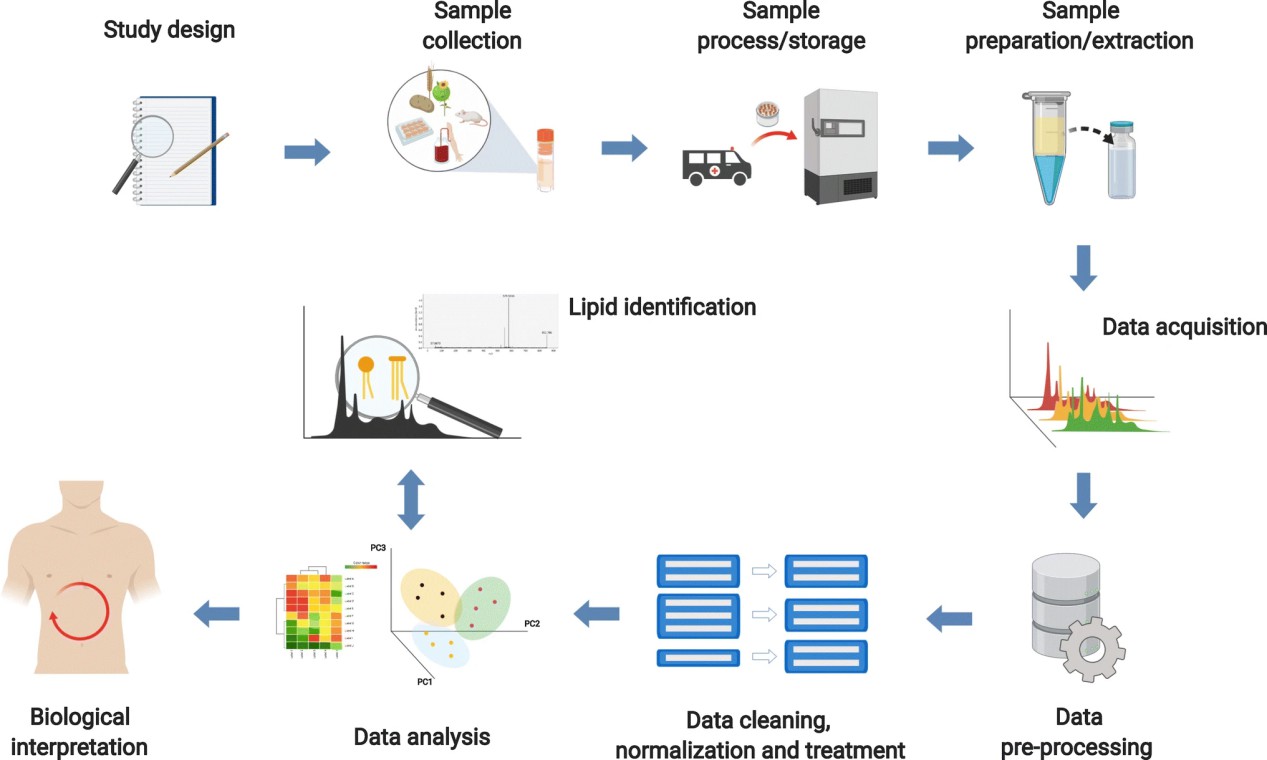 Figure 1. A typical workflow of lipidomics study. (Long N P, et al., 2020)
Figure 1. A typical workflow of lipidomics study. (Long N P, et al., 2020)
Acyl-Biotin Exchange (ABE) and Acyl-RAC
These chemical enrichment strategies are widely used to identify S-acylated (palmitoylated) proteins. The workflow proceeds in three main steps:
(1) Blocking of Free Thiols: Proteins are treated with N-ethylmaleimide (NEM) to irreversibly cap free cysteines.
(2) Cleavage of Thioester Bonds: Hydroxylamine (HAM) selectively cleaves palmitoyl thioester linkages, releasing the modified cysteine thiol.
(3) Biotin Labelling: Newly exposed thiols are biotinylated and captured using streptavidin beads.
Acyl-RAC (resin-assisted capture) replaces biotin with thiol-reactive resins. Both methods allow the enrichment of palmitoylated proteins, which are then analyzed by MS or immunoblotting.
Advantages: Enrichment improves detection sensitivity
Limitations: Indirect, does not distinguish lipid species
Bioorthogonal Chemical Reporters
This strategy uses lipid analogs with chemical handles such as azides or alkynes. These modified lipids are metabolically incorporated into proteins in living cells.
Subsequently, click chemistry reactions—typically copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC)—are used to conjugate fluorophores or affinity tags. This enables fluorescence imaging of lipidated proteins, affinity purification for MS analysis, and pulse-chase studies for lipidation dynamics. This approach allows for real-time visualization and quantification of lipidation under physiological conditions.
Advantages: Dynamic, non-radioactive, versatile
Limitations: Requires synthetic lipid analogs, possible off-target incorporation
Radioactive Metabolic Labelling
This classical approach involves culturing cells in the presence of radioactively labeled lipid precursors such as [³H]-myristic acid, [³H]-palmitic acid, or [¹⁴C]-mevalonate. Lipid-modified proteins incorporate these tracers during biosynthesis.
Following labeling, proteins are separated by SDS-PAGE and visualized via fluorography or autoradiography. This technique allows direct detection of lipid incorporation but does not identify specific modification sites. It also poses safety concerns due to radiation exposure. Additionally, the method lacks the resolution and throughput of modern analytical platforms.
Advantages: High specificity, suitable for kinetic studies
Limitations: Radioactive waste, low sensitivity, poor site resolution
Advanced Omics and High-Throughput Strategies
The study of protein lipidation has evolved beyond traditional biochemical methods, embracing cutting-edge omics and high-throughput strategies. These advanced approaches offer unparalleled sensitivity, scalability, and multidimensional insights into the lipidation landscape under various biological conditions.
Lipidomics-Proteomics Integration
Integrated lipidomics and proteomics analyses allow researchers to investigate how specific lipid species influence or correlate with protein lipidation. Lipidomics characterizes the entire lipid repertoire within a biological system using ultra-high-performance liquid chromatography coupled with tandem mass spectrometry (UHPLC-MS/MS). When this data is combined with proteomic datasets, it enables the identification of lipid-protein pairs and the elucidation of regulatory networks involved in membrane dynamics and signal transduction.
This integration also facilitates the identification of lipid metabolism alterations that may drive aberrant lipidation in pathological conditions such as cancer or metabolic disorders.
Quantitative Mass Spectrometry-Based Techniques
Tandem Mass Tagging (TMT): This isobaric labeling method enables multiplexed quantification of lipidated proteins across different biological samples. TMT tags do not interfere with peptide fragmentation, ensuring accurate quantification and allowing comparative studies of lipidation under varied stimuli or treatment conditions.
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC): SILAC introduces isotopically labeled amino acids into cells, enabling relative quantification of proteins in lipid-modified and unmodified states. When combined with lipid analog enrichment strategies, SILAC-MS can monitor temporal changes in lipidation during cellular signaling events.
Label-Free Quantification (LFQ): In situations where metabolic labeling is impractical, LFQ offers a robust alternative by comparing MS signal intensities of lipidated peptides across samples, providing high-throughput quantification without chemical modification.
Select Service
Chemical Proteomics with Bioorthogonal Lipid Probes
Chemical proteomics employs synthetic lipid analogs that mimic natural lipid moieties but contain clickable chemical handles (e.g., azide or alkyne groups). These probes can be metabolically incorporated into lipidated proteins within living cells.
Following incorporation, bioorthogonal chemistry—most commonly copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC)—facilitates the conjugation of fluorescent dyes or affinity tags to the lipidated proteins. This enables visualization via fluorescence microscopy, enrichment using streptavidin or other affinity-based pull-down methods, and identification and site mapping via mass spectrometry. This strategy is widely used to profile dynamic changes in palmitoylation, myristoylation, and prenylation with high specificity and minimal background noise.
Enrichment-Based Shotgun Proteomics
Shotgun proteomics using high-resolution MS instruments such as Orbitrap or Q-TOF systems provides deep coverage and accurate identification of enriched proteins.
Acyl-RAC (Resin-Assisted Capture) and ABE (Acyl-Biotin Exchange) are widely applied for S-palmitoylated proteins. These methods selectively cleave the thioester bond and replace it with a biotin or resin tag, allowing efficient purification.
Prenylated Protein Enrichment: Novel resins functionalized with thiol-reactive groups or synthetic ligands mimicking prenyl groups can selectively capture prenylated proteins for downstream analysis.
Bioinformatics Tools for Lipidation Prediction
Bioinformatics accelerates the identification of lipidation sites prior to empirical validation. Several specialized tools employ sequence motifs, machine learning, and structural features.
Motif-Based Predictors
Tools such as PrePS and iPreny-PseAAC use consensus sequences (e.g., "CAAX" box for prenylation). They scan protein termini for characteristic patterns. These methods achieve high specificity but may miss noncanonical sites.
Machine Learning Models
CSS-Palm and GPS-Lipid integrate amino acid properties, evolutionary conservation, and physicochemical descriptors. They employ support vector machines or gradient boosting. These models improve sensitivity and accommodate diverse species.
Deep Learning Approaches
Emerging platforms like DeepPalm apply convolutional neural networks to raw sequence windows. These frameworks capture nonlinear correlations and predict novel palmitoylation motifs without manual feature engineering.
Database Resources
SwissPalm, dbPTM, and LipidPTM curate experimentally verified lipidation events. They provide searchable entries with site annotations, enzyme associations, and cross-referenced literature. These repositories support training and benchmarking of predictors.
Structural Modeling
Tools such as ModPred map predicted sites onto three-dimensional protein models. Molecular dynamics simulations assess lipid-protein interactions and membrane affinity.
Applications of Protein Lipidation Analysis
Disease Biomarker Discovery
Altered lipidation patterns serve as biomarkers for cancer, infectious diseases, and neurodegeneration. In cancer, altered lipidation of signaling proteins contributes to tumorigenesis and metastasis. In neurodegenerative diseases, such as Alzheimer's and Parkinson's, disruptions in protein palmitoylation affect synaptic function and neuronal survival.
Case: Martin and colleagues utilized 17-octadecynoic acid (17-ODYA) for metabolic labeling combined with SILAC and pulse-chase approaches to quantitatively map dynamic protein palmitoylation events. This method distinguished stably palmitoylated proteins from those undergoing rapid turnover and highlighted enzymatic depalmitoylation as a regulatory mechanism for specific palmitoylation cycles (Martin et al., 2012).
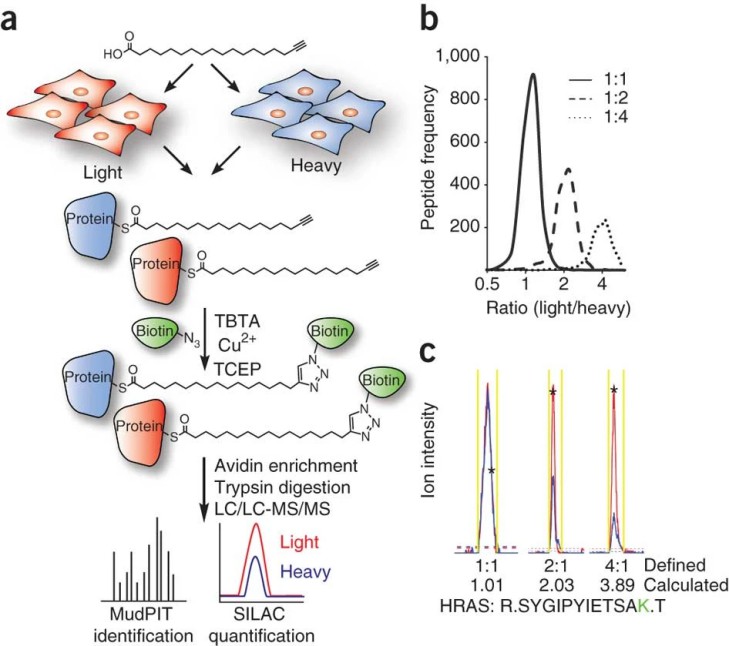 Figure 2. Quantitative analysis of protein palmitoylation.
Figure 2. Quantitative analysis of protein palmitoylation.
Drug Target Validation and Inhibitor Screening
Enzymes involved in protein lipidation pathways serve as potential therapeutic targets. Inhibiting these enzymes can modulate the lipidation status of proteins, thereby altering disease progression.
Case: Kho et al. introduced a "tagging-via-substrate" (TAS) strategy in which a synthetic azide-containing farnesyl substrate was metabolically incorporated into prenylated proteins. This enabled detection of 18 farnesylated proteins in a single experiment and facilitated inhibitor screening using streptavidin-based immunodetection (Kho et al., 2004).
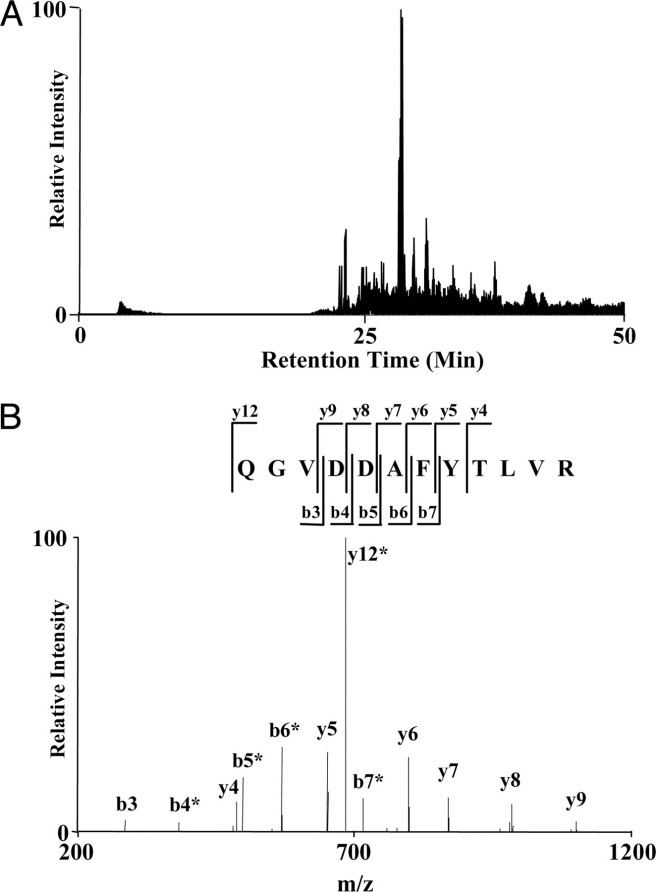 Figure 3. An example of nano-HPLC/MS/MS analysis for protein identification.
Figure 3. An example of nano-HPLC/MS/MS analysis for protein identification.
Drug Efficacy and Mechanism of Action Studies
Evaluation of how lipidation inhibitors (e.g., palmitoylation blockers) alter cancer cell viability and signaling networks.
Case: Metabolic labeling with clickable lipids, quantitative LC-MS to measure global palmitoylation changes and cell‐based functional assays. Advances in chemical proteomic profiling reveal how small-molecule inhibitors perturb palmitoylation networks, impacting tumor cell survival (Do-Won Jeong et al., 2023).
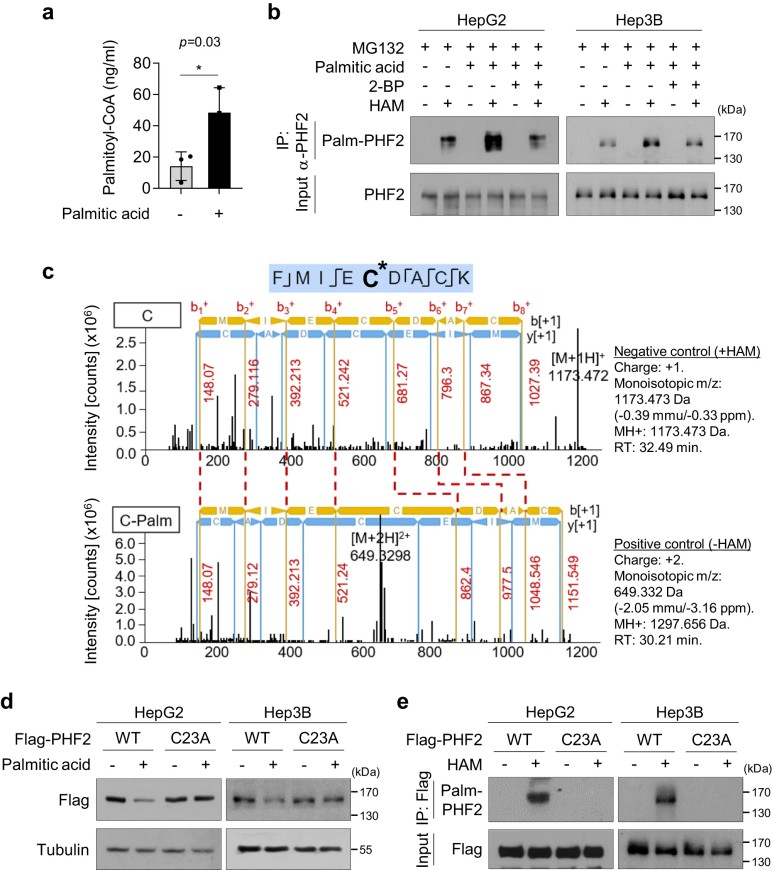 Figure 4. PHF2 is palmitoylated at cysteine 23.
Figure 4. PHF2 is palmitoylated at cysteine 23.
References
- Xu M, Xu B. Protein lipidation in the tumor microenvironment: enzymology, signaling pathways, and therapeutics. Molecular Cancer, 2025, 24(1): 1-50. DOI: 10.1186/s12943-025-02309-7
- Triola Long N P, et al. Advances in liquid chromatography–mass spectrometry-based lipidomics: a look ahead. Journal of Analysis and Testing, 2020, 4: 183-197. DOI: 10.1007/s41664-020-00135-y
- Fhu C W, Ali A. Protein lipidation by palmitoylation and myristoylation in cancer. Frontiers in Cell and Developmental Biology, 2021, 9: 673647. DOI: 10.3389/fcell.2021.673647
Related Articals

Our products and services are for research use only.
