Representing the expression, post-translational modification, and protein-protein interaction for certain cells, tissues, or organs, proteomics has become an increasingly important means to understand the molecular mechanism and signal transduction of human disease and cancer. Protein levels in the body are in a steady state of equilibrium. Accumulation of proteins usually leads to qualitative change in organism, thus causing tumorigenesis or disease. Therefore, quantification of proteins is significant for mechanism investigation. Herein, we provide an overview of the currently popular protein quantification technologies and their application in clinical research.
Introduction of HPLC-MS/MS-based Proteomics Technology
The development of high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) based proteomics technology mainly depends on the liquid chromatography-mass spectrometry equipment, sample separation and preparation technology, identification, as well as bioinformatics. At present, shotgun method is the most used large-scale protein identification strategy. As showed in figure1a, in the bottom-up approach, proteins are digested into a mixture of peptides by proteolytic enzymes, then separated and simplified by chromatography or affinity separation techniques, followed by the detection of segment ions via mass spectrometry. A search engine is engaged to match the mass spectrum with the spectrum generated by protein theoretical digestion in the database, and scored based on a series of indicators to achieve peptide-spectrum match (PSMs), and finally complete proteome identification. While in top-down mass spectrometry (TD-MS)-based proteomics,intact proteins are directly separated and detected from the samples. As compared with bottom-up approaches, TD-MS is capable of providing more information on preserved protein species. These two main strategies facilitate the proteomics research of diseases and cancers.
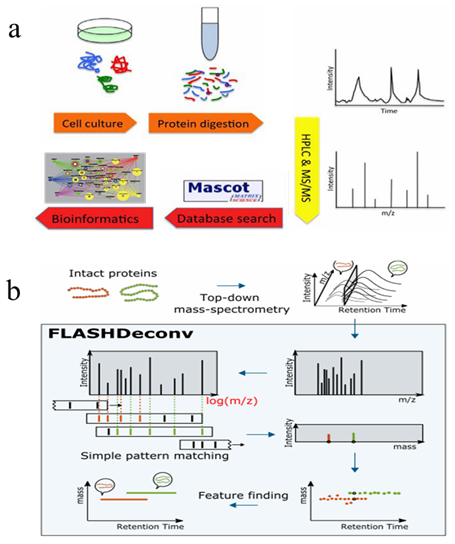
Figure 1. Workflow of bottom-up (a) and Top-down (b) mass spectrometry-based proteomics (Jeong K, et al, 2020)
Commonly-used Proteomics Quantitative Techniques and Their Clinical Application
Label-free analytical technique is the most widely used proteome quantification strategy because of its simplicity and minimal invasiveness. No additional biomolecules are involved. It is convenient and economic. However, researchers can only obtain the relative quantity of the proteins through LFQ method. LFQ is frequently used to search tumor biomarkers in clinical studies, as tumor adjacent or normal tissues are often treated as control group. de Oliveira et al. found that MMP-7 was presented as a more promising biomarker associated with useful assays of clinical practice for gastric adenocarcinoma patient diagnosis via LFQ results. Urine E-cadherin was supposed to be a marker for early detection of kidney injury in diabetic patients. In Sun’s research, label-free quantification was applied to systematically compare the protein profiling in saliva and serum exosomes from healthy subjects and lung cancer patients, and potential candidates were discovered as biomarkers for lung cancer. Dermcidin, apolipoprotein D, clusterin, prolactin-inducible protein and serum albumin are the most abundant sweat secreted proteins by targeted proteomics and label-free quantification mass spectrometry, providing potential sweat biomarkers which form the chemical barrier of the skin. Moreover, LFQ is commonly used in biomarker detection of other diseases and tumors, such as Vps35 for Parkinson's disease, SOAT1 for hepatocellular carcinoma, GOT2 for prostate cancer, A1AT for bladder cancer.
Stable isotope labeling with amino acids in cell culture is an in vivo metabolic labeling method, which is highly accurate and reliable for both absolute and relative protein quantification. The isotope of Lys (13C or 15N) and Arg (13C or 15N) are added to the cell medium lacking Lys and Arg, labelling proteins as light-type and heavy-type. Quantification are launched through MS1 level detected by mass spectrometry (figure 2). Fricke et al used SILAC to quantify the extracellular vesicles’ expression level in TGFBR2 deficiency colorectal cancers, uncovered 48 TGFBR2-regulated proteins. SILAC strategy is often used to identify post-translational modification (PTM) of proteins and amino acids. For example, Peng et al found that overexpression of RNF38 facilitates TGF-β signaling by ubiquitinating and degrading AHNAK in hepatocellular carcinoma. Zhang et al compared histone H3 and H4 post- translational modifications of esophageal squamous cell carcinoma with different invasive capabilities via SILAC quantification, and their studies contribute to understanding the different expressions of global histone PTMs in different invasive ESCC cell lines. With the development of super-SILAC technology, SILAC-based quantitative proteomics of tissues from model organisms are achieved by labeling whole organisms and using their tissues as internal standards, which elucidated the application of SILAC for clinical research. The super-SILAC is used for relative quantification of the clinical samples to find individual biomarkers.
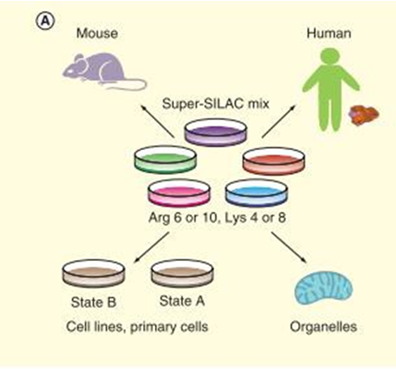
Figure 2. Workflow of SILAC (Shenoy A, et al, 2015).
The isobaric tags for relative and absolute quantification technology were developed by American ABI Company in 2004. It can label up to 8 samples at once depends on the iTRAQ kit, which contains reporter group, peptide reactive group and balance group. The molecular weight of reporter group ranging from 113Da to 121Da, and the related balance group ranging from 32Da to 24Da, which makes the total molecular weight to be 145Da. iTRAQ technology can compare proteins in many different samples with good reproducibility and high sensitivity. Qualitative and quantitative analyses can be performed simultaneously. This technology has been increasingly used in proteomics research. It has been successfully applied in the field of medical research, and can analyze and identify differential proteins in tumor tissues to find biomarkers. Hjelle Sigrun outlined the accurate knowledge of the molecular therapeutic targets in myeloid leukemia by iTRAQ. Hu et al identified two markers, elevated membrane- bound catechol-O-methyltransferase (MB-COMT) and attenuated retinol binding protein 4 (RBP4) in hepatic injury.
In order to quantify more protein samples, the tandem mass tags (TMT) labeling was developed. TMT can quantify up to 16 samples simultaneously. As showed in figure 3, TMT kit also contains reporter, normalizer, and amine-reactive group. TMT is designed to further increase the number of multiplex samples without sacrificing the goals of protein identification and quantitative quality, and it is better in quantitative accuracy. TMT is widely used in clinical sample research. Wu et al applied TMT to label samples from 10 HCC patients, and found plasminogen as a prognostic biomarker for HBV-related acute-on-chronic liver failure. Similar strategy is also applied in the biomarker discovery of gastric cancer, glioma, and glioblastoma.
Besides these commonly used methods above, other strategies such as difference gel electrophoresis (DIGE), Isotope-coded affinity tag (iCAT), and sequential windowed acquisition of all theoretical fragment ions (SWATH) can also be used to quantify proteins. These methods are also used to study molecular mechanisms and biomarkers.
Protein quantification technologies are essential in finding biomarkers for human cancers, drug targets selection, and mechanism studies. Some label-free and labeled methods are listed in table 1. These technologies are powerful tools that can help to study the mechanism of disease occurrence and progression of tumors, discovering new biomarkers, and elucidating the clinical research.
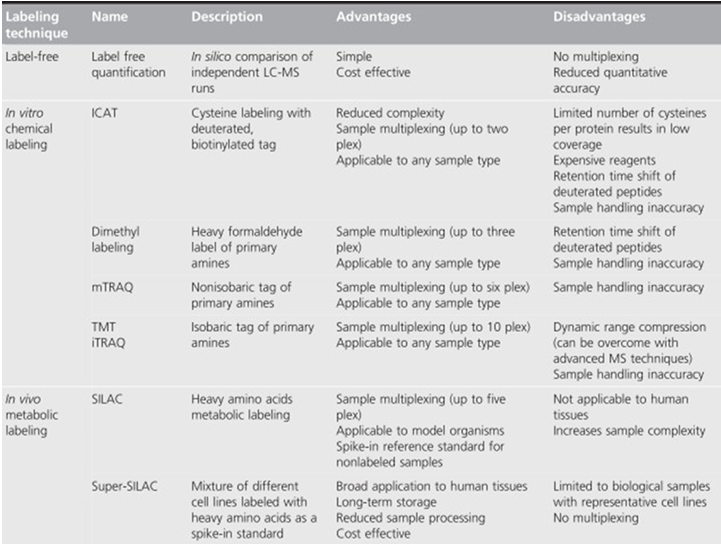
Table1 Advantages and disadvantages of mass spectrometry-based proteomics quantification. (Shenoy A, et al, 2015)
References
- Prianichnikov N, Koch H, Koch S, et al. MaxQuant software for ion mobility enhanced shotgun proteomics. Mol Cell Proteomics. 2020.
- Jeong K, Kim J, Gaikwad M, et al. FLASHDeconv: Ultrafast, High-Quality Feature Deconvolution for Top-Down Proteomics. Cell Syst. 2020;10: 213-218 e216.
- Chen D, Lubeckyj RA, Yang Z, et al. Predicting Electrophoretic Mobility of Proteoforms for Large-Scale Top-Down Proteomics. Anal Chem. 2020;92: 3503-3507.
- Soltermann F, Foley E, Pagnoni V, et al. Quantifying protein-protein interactions by molecular counting with mass photometry. Angew Chem Int Ed Engl. 2020.
- Hogrebe A, von Stechow L, Bekker-Jensen DB, Weinert BT, Kelstrup CD, Olsen JV. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat Commun. 2018;9: 1045.
- Wang X, Shen S, Rasam SS, Qu J. MS1 ion current-based quantitative proteomics: A promising solution for reliable analysis of large biological cohorts. Mass Spectrom Rev. 2019;38: 461-482.
- de Oliveira TM, de Lacerda J, Leite GGF, et al. Label-free peptide quantification coupled with in silico mapping of proteases for identification of potential serum biomarkers in gastric adenocarcinoma patients. Clin Biochem. 2020.
- Koziolek M, Mueller GA, Dihazi GH, et al. Urine E-cadherin: A Marker for Early Detection of Kidney Injury in Diabetic Patients. J Clin Med. 2020;9.
- Sun Y, Liu S, Qiao Z, et al. Systematic comparison of exosomal proteomes from human saliva and serum for the detection of lung cancer. Anal Chim Acta. 2017;982: 84-95.
- Csosz E, Emri G, Kallo G, Tsaprailis G, Tozser J. Highly abundant defense proteins in human sweat as revealed by targeted proteomics and label-free quantification mass spectrometry. J Eur Acad Dermatol Venereol. 2015;29: 2024-2031.
- Martinez A, Lectez B, Ramirez J, et al. Quantitative proteomic analysis of Parkin substrates in Drosophila neurons. Mol Neurodegener. 2017;12: 29.
- Jiang Y, Sun A, Zhao Y, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567: 257-261.
- Quanico J, Franck J, Gimeno JP, et al. Parafilm-assisted microdissection: a sampling method for mass spectrometry-based identification of differentially expressed prostate cancer protein biomarkers. Chem Commun (Camb). 2015;51: 4564-4567.
- Yang N, Feng S, Shedden K, et al. Urinary glycoprotein biomarker discovery for bladder cancer detection using LC/MS-MS and label-free quantification. Clin Cancer Res. 2011;17: 3349-3359.
- Choi Y, Jeong K, Shin S, et al. MS1-level proteome quantification platform allowing maximally increased multiplexity for SILAC and in vitro chemical labeling. Anal Chem. 2020.
- Fricke F, Michalak M, Warnken U, et al. SILAC-Based Quantification of TGFBR2-Regulated Protein Expression in Extracellular Vesicles of Microsatellite Unstable Colorectal Cancers. Int J Mol Sci. 2019;20.
- Lee FT, Mountain AJ, Kelly MP, et al. Enhanced efficacy of radioimmunotherapy with 90Y- CHX-A''-DTPA-hu3S193 by inhibition of epidermal growth factor receptor (EGFR) signaling with EGFR tyrosine kinase inhibitor AG1478. Clin Cancer Res. 2005;11: 7080s-7086s.
- Zhang K, Li L, Zhu M, et al. Comparative analysis of histone H3 and H4 post-translational modifications of esophageal squamous cell carcinoma with different invasive capabilities. J Proteomics. 2015;112: 180-189.
- Shenoy A, Geiger T. Super-SILAC: current trends and future perspectives. Expert Rev Proteomics. 2015;12: 13-19.
- Saraon P, Cretu D, Musrap N, et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol Cell Proteomics. 2013;12: 1589-1601.
- Liang S, Xu Z, Xu X, Zhao X, Huang C, Wei Y. Quantitative proteomics for cancer biomarker discovery. Comb Chem High Throughput Screen. 2012;15: 221-231.
- Bantscheff M, Eberhard D, Abraham Y, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25: 1035-1044.
- Hjelle SM, Forthun RB, Haaland I, et al. Clinical proteomics of myeloid leukemia. Genome Med. 2010;2: 41.
- Hu Z, Lausted C, Yoo H, et al. Quantitative liver-specific protein fingerprint in blood: a signature for hepatotoxicity. Theranostics. 2014;4: 215-228.



