- Service Details
- FAQ
What is PTMs?
The genetic information encoded in the DNA sequence is transcribed into mRNA and subsequently translated into proteins with specific amino acid sequences. However, the incorporation of post-translation modifications (PTMs) into proteins significantly enhances the functional diversity of the proteome. In complex mammalian cells, the genome comprises approximately 25,000 genes. Following transcription, the transcriptome encompasses over 100,000 transcripts. Through translation, the proteome consists of more than 100,000 proteins. Subsequently, PTMs and splicing recombination generate over 1,000,000 distinct protein species. It should be noted that apart from changing the properties of a protein through proteolytic cleavage, many translated proteins exhibit suboptimal activity within organisms and necessitate chemical modifications for full functionality. These modifications, known as PTMs, are essential for the attainment of a fully active protein state.
PTMs refers to the covalent processing events that occur during protein biochemical synthesis. PTMs processes mainly include: 1) the addition of chemical functional groups such as acylation, alkylation, phosphorylation, and glycosylation; 2) the attachment of protein or polypeptides, such as SUMOylation and ubiquitination; 3) structural changes in proteins such as disulfide bond formation. Adding or changing specific chemical functional groups on specific amino acids in a protein's amino acid sequence will not only affect the folding process and structure of the protein, but also has the potential to generate proteins with entirely distinct functionalities. Up to now, the reports indicate that there are over 200 PTMs, encompassing all types of proteins including nuclear transcription factors, metabolic enzymes, structural proteins, and plasma-membrane receptors, which demonstrates the diversity of PTMs.
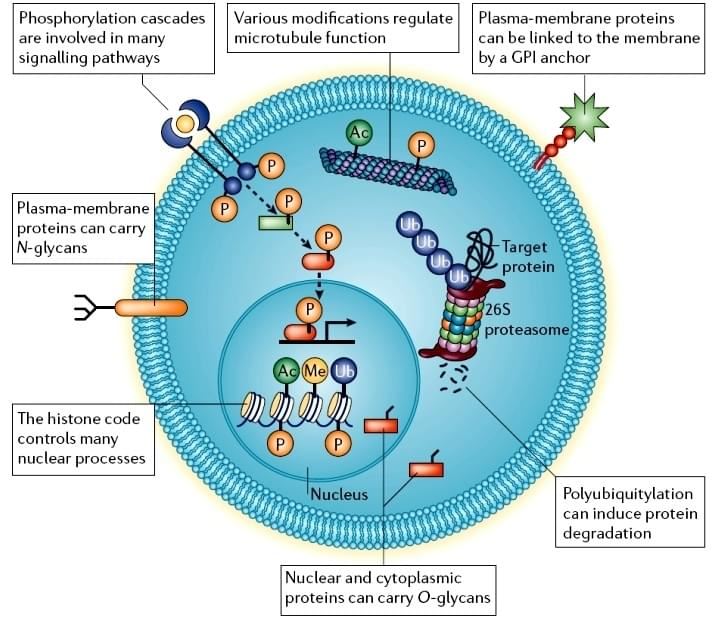 Figure 1. Cellular post-translational modifications [1].
Figure 1. Cellular post-translational modifications [1].
MS-Based Analysis of PTMs.
The regulation of numerous vital life processes in organisms relies not only on the relative abundance of proteins, but also on a diverse array of spatiotemporally specific and reversible PTMs. Therefore, elucidating the occurrence rules of PTMs is an important prerequisite for comprehending the intricate and diverse biological functions exhibited by proteins. With the development of sensitive high-resolution mass spectrometry (MS) and of highly sophisticated search algorithms and bioinformatic tools, enable accurately determine the molecular mass of proteins or peptides, and the molecular mass of a protein will change accordingly after it is PTM modified, based on this characteristic, the MS technique is applied to the identification of protein PTMs. The MS-based proteomics has made significant advancements in the past decade, enabling comprehensive identify and quantitative analysis of the proteome, encompassing PTMs, protein-protein interactions, and subcellular localization. Recently, MS-based studies have identified hundreds of thousands of PTM sites and found many new PTMs in mammalian proteome. Given the widespread occurrence, low stoichiometry and rapid dynamics of PTMs, PTM proteins or peptides must be enriched before MS identification and quantification.
Table 1. Some common and important post-translational modifications [2].
| PTM types | ΔMass (Da) | Stabilityb | Function and notes |
|---|---|---|---|
| Phosphorylation pTyr pSer,pThr | +80 +80 | +++ +/++ | Reversible, activation/inactivation of enzyme activity, modulation of molecular interactions, signaling |
| Acetylation | +42 | +++ | Protein stability, protection of N terminus. Regulation of protein-DNA interactions (histones) |
| Methylation | +14 | +++ | Regulation of gene expression |
| Acylation, fatty acid modification Farnesyl Myristoyl Palmitoyl etc. | +204 +210 +238 | +++ +++ +/++ | Cellular localization and targeting signals, membrane tethering, mediator of protein-protein interactions |
| Glycosylation N-linked O-linked | >800 203, >800 | +/++ +/++ | Excreted proteins, cell-cell recognition/signaling O-GlcNAc, reversible, regulatory functions |
| GPI anchor | >1,000 | ++ | Glycosylphosphatidylinositol (GPI) anchor. Membrane tethering of enzymes and receptors mainly to outer leaflet of plasma membrane |
| Hydroxyproline | +16 | +++ | Protein stability and protein-ligand interactions |
| Sulfation (sTyr) | +80 | + | Modulator of protein-protein and receptor-ligand interactions |
| Disulfide bond formation | -2 | ++ | Intra- and intermolecular crosslink, protein stability |
| Deamidation | +1 | +++ | Possible regulator of protein-ligand and protein-protein interactions, also a common chemical artifact |
| Pyroglutamic | -17 | +++ | Protein stability, blocked N terminus |
| Ubiquitination | >1,000 | +/++ | Destruction signal. After typic digestion, ubiquitination site is modified with the Gly-Gly dipeptide. |
| Nitration of tyrosine | +45 | +/++ | Oxidation damage during inflammation |
bStability: + labile in tandem mass spectrometry, ++ moderately stable, +++ stable.
The basic workflow of PTMs identification and quantification.
The fundamental workflow is as follows:
- The experiment designed (clear intention);
- Choice of MS instrument and analysis method;
- Methods optimization;
- Sample preparation-protein extraction and determination of concentration;
- Proper protease(s) for full proteolytic digestion;
- PTM-peptides enrichment (optional);
- Peptides fractionations for in-depth identification by reversed phase high performance liquid chromatography (optional);
- LC-MS/MS analysis;
- Bioinformation analysis for full PTM proteins and sites annotation.
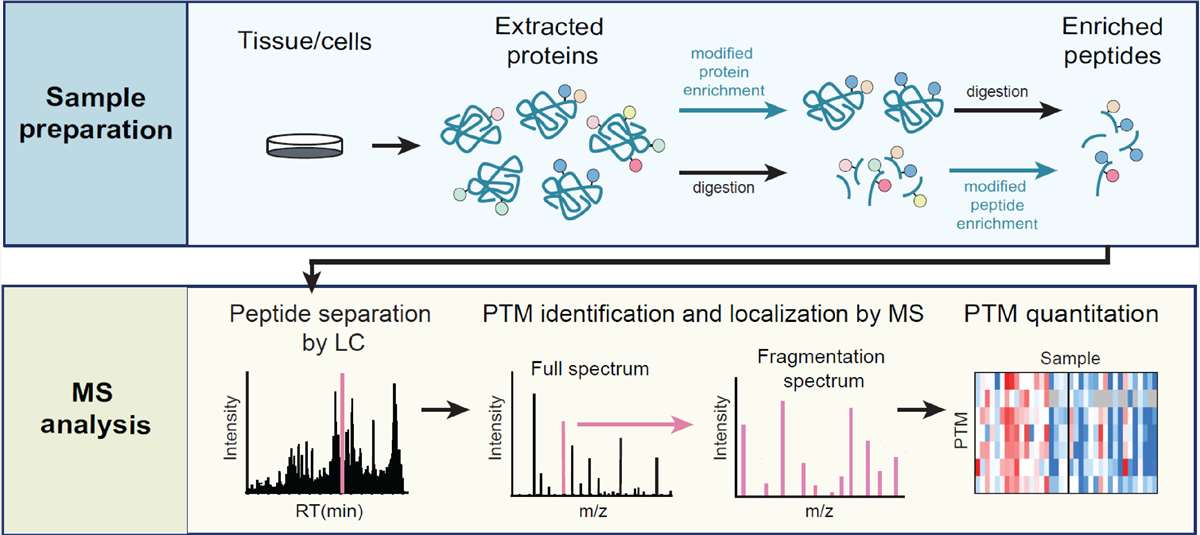 Figure 2. Mass spectrometry-based workflow for the identification of PTMs [3].
Figure 2. Mass spectrometry-based workflow for the identification of PTMs [3].
The table 2 showcases a wide range of enrichment strategies for PTM peptides.
Table 2. Enrichment strategies for MS-based PTM proteomic analysis [3].
| Protein PTMs | Target amino acid residues | Affinity enrichment strategies | Chemical enrichment strategies |
|---|---|---|---|
| Phosphorylation | Ser, Thr, Tyr, His, Arg, Lys, Asp, Cys, Glu | IMAC, MOAC, SIMAC, SIMAC-HILIC, SCX, SAX, immunoenrichment, superbinder SH2 domains, ERLIC, hydroxyapatite AC, HILIC-IMAC | DBHA probe, HA-yne probe, isoDTB tag, photo-pTyr-scaffold probe, sulfonyl-triazole probe |
| Acetylation | Lys, Ser, Thr, N terminus | immunoenrichment, SCX, ZIC-HILIC, COFRADIC, antibody-IEF, antibody-SCX | alkyne-containing thioester probe, metabolic labelling by ethyl fluoroacetate |
| Methylation | Lys, Arg, His, Ala, Asn | immunoenrichment, IEF, SCX, HILIC, antibody-SCX, antibody-HpH RP, 3xMBT methyl-binding domains, antibody-propionylation | azide- and alkyne-analogues of SAM, chemoenzymatic labelling by ProSeAM |
| Glycosylation | Asn, Arg (N-linked) Ser, Thr, Tyr (O-linked) | lectins, modified glycosidases, immunoenrichment, IMAC, MOAC, HILIC | metabolic labelling by: Ac4ManNAz, Ac4GalNAz, Ac4FucAl, Ac4ManNAl, GalNAz, 1,3-Pr2GalNAz, GalNAzMe; chemoenzymatic labelling by UDP-GalNAz, Glyco-TQ |
| Ubiquitination | Lys, N terminus, Cys, Ser, Thr | protein degradation, trafficking, regulation of enzymes, translation, DNA repair | tagged-Ub, immunoenrichment, COFRADIC |
| Sumoylation | Lys | protein interactions, subcellular localization, enzymatic activities | tagged-SUMO, immunoenrichment, SUMO-interacting motifs |
IMAC: immobilized metal ion affinity chromatography; MOAC: metal oxide affinity chromatography; HILIC: hydrophilic interaction liquid chromatography; SAX: strong anionic exchanges; COFRADIC: combined fractional diagonal chromatography; DBHA: desthiobiotin containing hydroxylamine; ERLIC: electrostatic repulsion liquid interaction chromatography.
With years' experience in advanced experiment equipment, Creative Proteomics can provide a variety of PTM services to assist your scientific research, including:
Furthermore, we excel in providing PTM quantitative proteomics services as well, particularly in SILAC, iTRAQ/TMT, label-free, DIA and target proteomics (SRM/PRM). All of these technologies facilitate the analysis and interpretation of PTM. For comprehensive information on protein PTMs, please refer to the article "Overview of Post-translational Modification Analysis".
The Advantages of MS-based PTMs service on Creative Proteomics.
- Highly accurate and reliable quantification results: The utilization of advanced MS technology, well-established protein quantification methods, along with integration of PTM protein or peptide enrichment strategies, ensures precise and high-confidence identification and quantification results of PTMs (with a variation coefficient <5%).
- High applicability of various sample types: Archaea, prokaryotes, cells, tissues, and bodily fluids, etc.
- Professional detection and analysis capability: Experienced PTMs identification and quantification technical team, strict quality control system, together with ultra-high resolution detection system and proficient data pre-processing and analysis capability, ensures the delivery of reliable and accurate data.
- Reproducible: Obtain consistent and reproducible inter- and intra- assay results for data analysis.
- High throughput and sensitivity: Q-Exactive, Q-Exactive HF, Orbitrap Fusion™ Tribrid™, etc.
- The results are delivered in a short turnaround with high quality.
How to place an order
With over 10 years experimental experience, Creative Proteomics have the expertise to provide a tailored and scientific experimental scheme that meets your specific requirements for conducting PTMs analysis, facilitating precise and accurate identification and quantification of PTM-sites and -proteins. Please feel free to contact us by email to discuss your specific needs. Our customer service representatives are available 24 hours a day, from Monday to Sunday.

References
- Jensen ON. Interpreting the protein language using proteomics. Nature Reviews Molecular Cell Biology. 2006 Jun;7(6):391-403.
- Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nature Biotechnology. 2003 Mar;21(3):255-61.
- Brandi J, Noberini R, Bonaldi T, et al. Advances in enrichment methods for mass spectrometry-based proteomics analysis of post-translational modifications. Journal of Chromatography A. 2022 Aug 16;1678:463352.
Q: What are the common post-translation modifications and what are the differences?
A:
| Modification Type | Amino Acid Site | Enrichment Method | Application |
|---|---|---|---|
| Phosphorylation | Ser/Thr/Tyr | TiO2, IMAC-Fe, Sequence-specific antibodies | Signal transduction, cell cycle, regulatory mechanisms, stress response, growth and development, and cancer mechanisms, etc. |
| Acetylation | Lys | Sequence-specific antibodies | Gene expression regulation, cell defense mechanisms, cell apoptosis and metabolism, cell cycle, transcriptional activation and silencing, protein stability, and neurodegenerative diseases, etc. |
| Ubiquitination | Lys | Sequence-specific antibodies | Cell cycle, cell apoptosis, protein degradation, defense mechanisms, photomorphogenesis, signal transduction, plant growth and development, cancer, and neurodegenerative diseases, etc. |
| Succinylation | Lys | Sequence-specific antibodies | Biosynthesis and metabolism, inflammation and disease, central metabolic pathways and epigenetic regulation |
| Methylation | Arg/Lys | Sequence-specific antibodies | Epigenetics, cancer mechanisms, aging, neurodegenerative diseases, protein translocation and signal transduction, and histone function, etc. |
| N-Glycosylation | Asn | HILIC | Cell recognition, differentiation, stress response, signal transduction, immune response, neurodegenerative diseases, metabolic diseases, and infectious diseases research, etc. |
| O-Glycosylation | Ser/Thr | HILIC |
Q: What are the differences between PTMs Omics and conventional quantitative proteomics?
A: In PTM proteomics, there is an additional process of enriching the modified peptide segment after enzymatic digestion. The identification of modification sites is included during data analysis. Differential analysis in PTM proteomics is based on the peptide level rather than the protein level.
Q: Why does the primary focus of PTM (Post-Translational Modification) proteomics lies in the quantitative analysis of modification sites rather than at the protein level?
A: This is attributed to the fact that a single protein may harbor multiple modification sites. Following enzymatic digestion, a majority of the resulting peptides comprise unmodified sequences, with a smaller fraction containing modified moieties.
Despite pre-enrichment of modified peptides prior to mass spectrometry analysis, the efficiency of enrichment may not reach 100%, and for certain types of modifications, the enrichment efficiency could be less than 50% or even lower. Additionally, non-modified peptides, detected during the analysis, contribute to protein quantification. The enrichment process often leads to the loss of a significant portion of non-modified peptides. Consequently, the quantified protein levels obtained do not accurately reflect the true protein expression levels within the biological system.
For label-free quantification of modifications, evaluating protein levels becomes inconsequential. The emphasis is rather placed on discerning the abundance differences in specific modification sites across different samples. This underscores why PTM proteomics primarily involves the quantitative analysis of modification sites.
Q: How to identify N-glycosylation sites?
A: Identification of N-glycosylation sites involves the following steps:
a) Protein digestion; b) Enrichment of N-glycosylated modified peptide segments; c) Use of glycosidases to cleave the glycan attached to ASN (asparagine) in 18O-labeled water, resulting in an increase of 2.9890 Da in the molecular weight of ASN; d) High-precision LC-MS mass spectrometry for detecting deglycosylated peptide segments; e) Database retrieval to confirm the changes in molecular weight after deglycosylation compared to the theoretical molecular weight and to determine the sequence of the glycosylated modified peptide segment, thereby pinpointing the glycosylation site on the protein.
Q: Can PRM be applied to the modified proteome?
A: In principle, PRM (Parallel Reaction Monitoring) can be utilized as long as there are internal standard peptide segments identified in previous omics analyses. However, it's important to note that PRM in the context of modified proteomics presents greater challenges and complexities.
Q: How are peptide segments with specific modifications enriched? Does the use of antibodies impact the results?
A: The enrichment methods for different modifications vary. Following the enrichment of peptide segments, there is a subsequent elution step during which the modified peptide segments are separated from the antibodies. This ensures that the results are not influenced by the presence of antibodies.
Q: How is the modified proteome analyzed?
A: Currently, the default approach involves using Label-free methods, and 4D (four-dimensional) proteomics can also be employed.
Q: What advantages does 4D proteomics offer compared to conventional proteomics?
A: 4D proteomics, based on the state-of-the-art timsTOF Pro mass spectrometer, utilizes Trapped Ion Mobility Spectrometry (TIMS) for quantitative proteomic studies. This technology introduces a fourth dimension, ion mobility, alongside the traditional dimensions of retention time, mass-to-charge ratio (m/z), and ion intensity. The inclusion of ion mobility significantly enhances scanning speed and detection sensitivity, leading to substantial improvements in identification depth, detection throughput, and quantitative accuracy in proteomic analysis.
Q: What are the applications of 4D proteomics?
A:
| Product | Technology | Application |
|---|---|---|
| 4D-PTMs | 4D-label free | Suitable for all sample types, particularly advantageous for trace samples like paraffin sections and biopsy tissues. Recommended for sample sets of up to 30 cases. |
| 4D-DIA | Requires a minimum of 6 samples, library construction is needed. Applicable for large-scale protein profiling analyses, such as clinical cohorts and population studies. Recommended for sample sets of 30 cases or more. | |
| 4D-PTMs Proteomics | 4D-Phosphorylation | Widest range of applications, nearly applicable to all biological processes. Used in studies related to drug targets, tumor development, neurological diseases, and commonly employed in the investigation of signal transduction, cell cycle, regulatory mechanisms, stress response, growth and development, and cancer mechanisms, among others. |
| 4D-Acetylation | Metabolic regulation: Induces changes in metabolic enzyme activity and metabolic pathway alterations. Applicable in studies of energy metabolism regulation, metabolic syndrome, cardiovascular disease development, etc. | |
| 4D-Ubiquitination | Protein expression degradation regulation: Mediates protein degradation through the ubiquitin-proteasome pathway. Used in studies of aging and degenerative diseases, immune inflammation, tumor progression, etc. |





