How to Integrate DIA-MS Proteomics With Your RNA-seq Study
When Your RNA-seq is Ready—But Proteomics Falls Behind
RNA-seq has revolutionized large-scale biomedical research. With its high-throughput design and deep transcriptome coverage, it enables scientists to generate clean, comprehensive gene expression matrices across hundreds—or even thousands—of biological samples. Whether profiling tumors, patient-derived organoids, or longitudinal cohorts, RNA-seq reliably reveals transcriptional changes at scale.
But transcription alone rarely tells the whole story.
The biological outcomes that matter—drug response, pathway activation, phenotypic switching—are ultimately executed by proteins, not transcripts. And that's where many multi-omics studies face a critical obstacle: the proteomics layer fails to keep pace.
Most conventional proteomics workflows weren't designed for integration with large RNA-seq datasets:
- DDA (Data-Dependent Acquisition) introduces stochastic sampling, yielding >30% missing values across protein matrices.
- TMT (Tandem Mass Tag) methods max out at ~18 samples per batch. Scaling to larger cohorts introduces batch effects, ratio compression, and comparability issues.
The result? A transcriptome-rich, but proteome-poor dataset—one that limits integration, weakens biological conclusions, and risks overlooking functionally relevant mechanisms.
That's why more researchers are turning to Data-Independent Acquisition Mass Spectrometry (DIA-MS).
DIA-MS offers:
- Systematic, unbiased peptide detection
- Matrix-level completeness with low missingness
- Cohort-scale throughput matching RNA-seq
- Re-analyzable raw data for long-term value
At Creative Proteomics, we deploy DIA-MS not just as a platform, but as a multi-omics engine—designed to integrate seamlessly with your RNA-seq workflows and scale with your study design.
In this resource, we'll show how DIA-MS overcomes the traditional proteomics bottleneck—and how you can build a truly scalable, reproducible, and insight-rich multi-omics strategy.
To understand how DIA-MS improves consistency and data depth in large studies, see DIA vs DDA Mass Spectrometry: A Comprehensive Comparison and Differences Between DIA, TMT/iTRAQ, and Traditional Label-free. These comparisons outline how DIA achieves higher reproducibility and scalability across complex cohorts.
Why DIA-MS Is the Only Proteomics Approach Built to Scale With Transcriptomics
As RNA-seq continues to define the standard for high-throughput, low-missing-rate data across large cohorts, proteomics often struggles to match that level of scalability and consistency. This isn't just a question of throughput—it's a structural limitation built into many traditional proteomics workflows.
In large-cohort studies, researchers commonly encounter the following:
- DDA uses real-time intensity-based selection of top N peptides in each run. This stochastic sampling leads to poor reproducibility across samples and high matrix-level missingness (>30%), which directly undermines integrative analysis with transcriptomic data.
- TMT–based workflows allow multiplexing, but the number of available channels per batch is limited (typically 11–18). Scaling to hundreds of samples introduces batch effects and quantification distortion due to ratio compression, complicating cross-batch comparability.
DIA-MS overcomes these challenges by using a fundamentally different acquisition strategy. It systematically fragments all ions across predefined mass windows in every run—regardless of signal intensity.
To understand the practical differences between methods, the following comparison outlines how DIA-MS aligns with RNA-seq in structure, scale, and data integrity.
 Comparative overview of RNA-seq, DIA-MS, and DDA showing scalability, data completeness, and integration readiness for multi-omics studies.
Comparative overview of RNA-seq, DIA-MS, and DDA showing scalability, data completeness, and integration readiness for multi-omics studies.
Comparative Overview: RNA-seq vs DIA-MS vs DDA
| Feature | RNA-seq | DIA-MS | DDA |
| Acquisition Strategy | Unbiased sequencing of all transcripts | Systematic fragmentation of all peptides | Intensity-driven selection of top-N precursors |
| Scalability | 100s–1000s of samples with minimal batch effects | Scales linearly across hundreds of samples | Scalability limited by run-to-run variability |
| Data Completeness | Near-zero missingness | Low missingness (<5%), highly consistent matrices | High missingness (>30%) typical in cohort studies |
| Data Reusability | FASTQ files support re-analysis and new pipelines | DIA raw files can be re-mined with updated libraries | No data for peptides not selected during acquisition |
| Integration Readiness | Matrix-structured, sample-aligned data | Matrix-structured, sample-aligned data | Requires heavy imputation; alignment to transcriptome is poor |
For deeper insights into workflow design and data reliability, explore Avoiding Failure in DIA Proteomics: Common Pitfalls and How to Fix Them and Library-Free vs Library-Based DIA Proteomics.
Designing RNA-seq + DIA-MS Integration: A Practical Study Framework
In multi-omics research, transcriptomics often serves as the starting point. Many teams already possess RNA-seq data from bulk tissue, cell models, or patient cohorts—but the transcriptome alone rarely explains the full functional landscape. To move from expression patterns to biological mechanisms, a matched proteomics layer is essential.
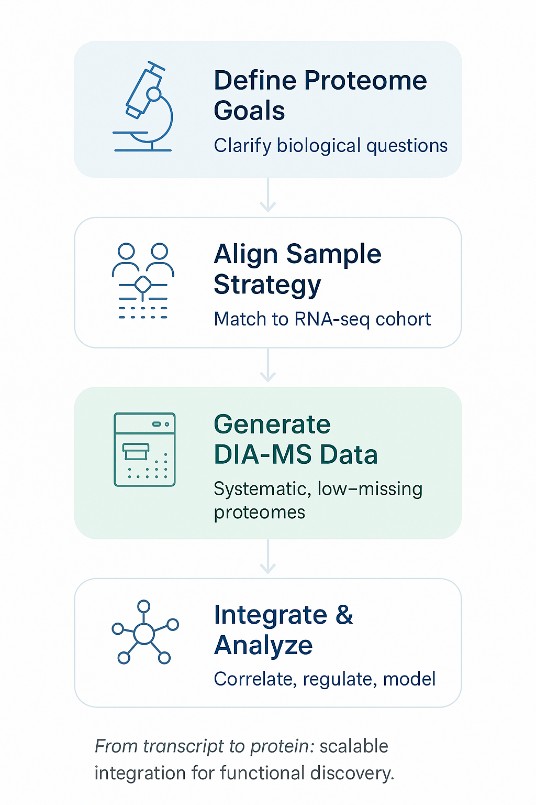
Below is a practical, hypothesis-driven study design framework for researchers looking to extend their transcriptomic insights using DIA-MS.
Step 1: Define the Analytical Goals for Proteome Layer
Before collecting proteomics data, clarify how it complements your RNA-seq results:
- Do transcript changes fail to explain phenotype or treatment response?
- Are you exploring suspected post-transcriptional regulation (e.g., differential translation, degradation, PTM activity)?
- Is your aim to uncover functional pathways or build predictive models beyond gene expression?
These questions guide sample selection, acquisition depth, and analytical focus on the proteomics side.
Step 2: Align Sample Strategy to Existing RNA-seq Cohort
To enable cross-omics integration:
- Use the same biological material or matched replicates as in the RNA-seq study whenever possible.
- For archived tissues or biofluids, ensure clear metadata traceability: sample ID, timepoint, treatment, clinical features.
- Document extraction protocols and storage conditions to assess potential batch effects.
If RNA-seq data comes from collaborators or external platforms, request metadata that enables proteomics samples to be matched appropriately.
Step 3: Generate a High-Quality DIA-MS Dataset
Once matched samples are selected, proteome profiling can begin using DIA-MS—a method well-suited for large-scale, quantitative multi-omics integration:
- Systematic acquisition (no stochastic sampling) ensures data consistency across samples.
- Low missingness rates (<5%) enable matrix alignment with transcriptome data.
- Optional PTM layers (e.g., phosphoproteomics) can capture regulation beyond abundance.
QC metrics such as protein ID count, peptide CV, and retention-time drift are recorded per run and per batch to support reproducibility.
Step 4: Perform Stepwise Integration Analysis
We recommend organizing the analysis across three tiers:
- Tier 1 – Descriptive Correlation
Quantify transcript–protein relationships across genes and pathways. Identify globally concordant modules and outliers.
- Tier 2 – Post-Transcriptional Regulation
Detect genes with stable mRNA but changing proteins (e.g., due to degradation or PTMs), or vice versa (e.g., translational buffering). This layer captures regulatory mechanisms not evident from RNA-seq alone.
- Tier 3 – Systems-Level Integration
Use methods like GSEA, WGCNA, or MOFA+ to integrate both data layers. Highlight pathways under dual regulation, and identify key drivers or modules with divergent RNA–protein behavior.
 Stepwise framework illustrating how RNA-seq and DIA-MS integrate within a scalable multi-omics workflow.
Stepwise framework illustrating how RNA-seq and DIA-MS integrate within a scalable multi-omics workflow.
Validated Use Cases: DIA-MS for Scalable, Reproducible Proteomics
Breast cancer: proteotype subtyping and targets beyond transcript calls
A cohort of 96 breast tumors was profiled by SWATH/DIA to generate "proteotypes" and data-driven subtypes. The study showed that protein-level classification both overlaps with and refines transcript-based subtypes, highlighting kinase and cell-cycle proteins with therapeutic relevance that are not reliably inferred from mRNA alone. (Bouchal et al., 2019. DOI: https://doi.org/10.1016/j.celrep.2019.06.046).
For teams starting from RNA-seq clusters, DIA supplied a stable, sample-aligned protein matrix that sharpened subtype definitions and exposed protein targets for follow-up.
Pan-cancer cell lines (NCI-60): drug-response modeling benefits from DIA proteomes
Using pressure-cycling technology with SWATH/DIA, >3,000 proteins were consistently quantified across the NCI-60 cell lines. Integrating these proteomes with existing genomics, transcriptomics, and pharmacology improved drug-response modeling versus single-omic predictors and enabled pathway-level interpretation of sensitivity. (Guo et al., 2019. DOI: https://doi.org/10.1016/j.isci.2019.10.059).
When the end goal is predictive modeling (e.g., screening-guided candidates), DIA-level consistency across many samples yielded features that better track functional response than RNA alone.
Liver cancers (HCC vs. iCCA): DIA resolves tumor-type differences aligned with biology
A clinical study applied DIA-MS to tumor and adjacent tissues from hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA), identifying distinct proteomic programs—lipid-metabolism upregulation in HCC and extracellular-matrix signatures in iCCA—supporting differential mechanisms not captured by transcript trends alone. (Zhu et al., 2023. DOI: https://doi.org/10.1016/j.mcpro.2023.100604).
For cohorts with mixed tumor types, DIA provided matrix-complete protein readouts that separated clinically relevant biology and refined pathway hypotheses for each disease.
Lung adenocarcinoma: activity-guided DIA reveals regulation not visible at mRNA level
An activity-based chemical proteomics workflow coupled to SWATH/DIA quantified enzyme families directly in primary LUAD tissues. The work identified activity-level signatures associated with aggressive phenotypes that were not evident from transcript measurements, illustrating a practical route to mechanism-level biomarkers. (Sajić et al., 2025. DOI: https://doi.org/10.1038/s41467-025-59564-x).
When function (enzyme activity) is the endpoint, DIA's systematic acquisition supports robust quantification and retrospective mining alongside RNA-seq.
References
- Krasny L. & Huang P. H. (2021). "Data‑independent acquisition mass spectrometry (DIA‑MS) for proteomic applications in oncology." Molecular Omics.
- Lou, R., & Shui, W. (2024). Acquisition and analysis of DIA-based proteomic data: A comprehensive survey in 2023. Molecular & Cellular Proteomics, 23(2), 100712.
- Goetze S., van Drogen A. (2024). "Simultaneous targeted and discovery‑driven clinical proteotyping using hybrid‑PRM/DIA." Clinical Proteomics, 21, Article 26.
- Wang Y., Mamie L. T‑S., Chen L., Xu Y., Kuczler M. D., Cao L., Pienta K. J., Amend S. R., Zhang H. (2022). "Optimized data‑independent acquisition approach for proteomic analysis at single‑cell level." Clinical Proteomics, 19, Article 24.
- Fröhlich, K., Fahrner, M., Brombacher, E., Seredynska, A., Maldacker, M., Kreutz, C., ... & Schilling, O. (2024). "Data‑Independent Acquisition: A Milestone and Prospect in Clinical Mass Spectrometry. " Molecular & Cellular Proteomics, 23(8), 100800.




 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)